Проліа®
Реєстраційний номер: UA/12077/01/01
Імпортер: Амджен Європа Б.В.
Країна: НідерландиАдреса імпортера: Мінервум 7061, НЛ-4817 ЗК Бреда, Нiдерланди
Форма
розчин для ін'єкцій, 60 мг/мл по 1 мл розчину в скляному попередньо наповненому шприці з голкою, закритою ковпачком, із захисним пристроєм від випадкового уколу голкою; по 1 попередньо заповненому шприцу з захисним пристроєм в блістері; по 1 блістеру в кортонній коробці; по 1 мл розчину в скляному попередньо заповненому шприці з голкою, закритою ковпачком; по 1 попередньо заповненому шприцу в блістері або без блістера, поміщеному в картонну коробку; по 1 мл розчину в скляному флаконі; по 1 флакону в картонній коробці
Склад
1 мл розчину містить 60 мг деносумабу
Виробники препарату «Проліа®»
Країна: США
Адреса: Стейт Роуд 31, Кілометер 24.6, Джанкос, Пуерто-Ріко 00777, США
Країна: Нідерланди
Адреса: Мінервум 7061, 4817 ZK, Бреда, Нідерланди
Інструкція по застосуванню
для медичного застосування лікарського засобу
ПРОЛІА®
(PROLIA®)
Склад
діюча речовина: деносумаб;
1 мл розчину містить 60 мг деносумабу;
допоміжні речовини: кислота оцтова льодяна, натрію гідроксид, сорбіт (E 420), полісорбат 20 (лише у попередньо заповненому шприці), вода для ін'єкцій.
Лікарська форма. Розчин для ін'єкцій.
Основні фізико-хімічні властивості: майже прозорий, безбарвний або жовтуватий розчин, що практично не містить сторонніх часток.
Фармакотерапевтична група. Лікарські засоби для лікування захворювань кісток, інші лікарські засоби, що впливають на структуру та мінералізацію кісток. Код АТХ M05B X04.
Фармакологічні властивості.
Фармакодинаміка.
Деносумаб - це моноклональне антитіло людини (IgG2), вироблене на клітинній лінії ссавців (CHO) з допомогою технології рекомбінантної ДНК, мішенню для якого є RANKL, з яким препарат зв'язується з високою афінністю та специфічністю, запобігаючи активації його рецептора RANK на поверхні прекурсорів остеокластів та остеокластів. Запобігання взаємодії RANKL/RANK пригнічує утворення остеокластів, погіршує їх функціонування та життєздатність, таким чином зменшуючи резорбцію як трубчастих, так і губчастих кісток.
Фармакодинамічні ефекти
Лікування препаратом Проліа® швидко зменшує рівень кісткового ремоделювання, досягаючи найнижчого рівня сироваткового маркера резорбції кісток - С-телопептидів колагену 1 типу (CTX) (85 % зменшення) - через 3 дні. Знижений рівень СТХ та утримується протягом усього інтервалу між застосуванням доз. В кінці кожного періоду після введення дози препарату ефект зниження рівня CTX був частково послаблений: з максимального зниження більш ніж на 87 % до зниження приблизно більш ніж на 45 % (у діапазоні від 45 до 80 %), що відображає оборотність ефектів деносумабу відносно ремоделювання кісткової тканини після зниження сироваткового рівня препарату. При продовженні терапії ці ефекти зберігалися.
Маркери ремоделювання кісткової тканини загалом досягали рівнів, на яких вони були до початку лікування препаратом, протягом 9 місяців після введення останньої дози. Після повторного ініціювання лікування рівень інгібування CTX деносумабом був подібним до того, що спостерігався у пацієнтів, які вперше розпочали лікування цим препаратом.
Імуногенність
За даними клінічних досліджень під час застосування препарату Проліа® не спостерігалося вироблення нейтралізуючих антитіл. Менше 1 % пацієнтів, які лікувалися деносумабом не більше 5 років, мали позитивні тести (за даними чутливого імунологічного методу) на нейтралізуючі зв'язуючі антитіла без ознак впливу на фармакокінетику, токсичність або клінічний ефект.
Лікування остеопорозу у жінок у постменопаузальний період
Ефективність та безпека препарату Проліа®, який застосовували 1 раз кожні 6 місяців протягом 3 років, досліджувались у жінок у постменопаузальний період (7808 жінок у віці 60-91 рік, з яких 23,6 % мали поширені переломи хребців) з вихідними Т-показниками мінеральної щільності кісток (МЩК) в поперековому відділі хребта або тазостегновому зчленуванні між -2,5 і -4,0 і середньою абсолютною 10-річною ймовірністю перелому 18,60 % (децили: 7,9-32,4 %) щодо основних остеопоротичних переломів і 7,22 % (децили: 1,4-14,9 %) щодо перелому шийки стегна. Жінки з іншими захворюваннями або ті, хто лікувався препаратами, що можуть впливати на кістки, були виключені з дослідження. Жінки отримували добавки кальцію (принаймні 1000 мг) і вітаміну D (принаймні 400 МО) щодня.
Вплив на переломи хребців
Проліа® значно знижує ризик виникнення нових переломів хребців на 1, 2 і 3 році застосування препарату (р < 0,0001) (див. таблицю 1).
Таблиця 1. Вплив Проліа® на ризик нових переломів хребців
|
Період застосування |
Частка жінок з переломом (%) |
Зниження абсолютного ризику (%) (95 % ДІ) |
Зниження відносного ризику (%) (95 % ДІ) |
|
|
Плацебо, n =3D 3906 |
Проліа®, n =3D 3902 |
|||
|
0-1 рік |
2,2 |
0,9 |
1,4 (0,8, 1,9) |
61 (42, 74)** |
|
0-2 роки |
5,0 |
1,4 |
3,5 (2,7, 4,3) |
71 (61, 79)** |
|
0-3 роки |
7,2 |
2,3 |
4,8 (3,9, 5,8) |
68 (59, 74)* |
*p < 0,0001, **p < 0,0001 - розвідувальний аналіз
Вплив на переломи шийки стегна
Препарат Проліа® продемонстрував відносне зниження на 40 % (0,5 % зниження абсолютного ризику) ризику перелому шийки стегна протягом 3 років (р < 0,05). Частота переломів шийки стегна була 1,2 % в групі плацебо в порівнянні з 0,7 % в групі Проліа® через 3 роки.
При апостеріорному аналізі у жінок > 75 років зниження відносного ризику на 62 % спостерігалося при застосуванні препарату Проліа® (1,4 % зниження абсолютного ризику, р < 0,01).
Вплив на всі клінічні переломи
Препарат Проліа® значно знизив кількість переломів в усіх типів (див. таблицю 2).
Таблиця 2. Вплив Проліа® на ризик клінічних переломів протягом 3 років
|
Типи переломів |
Частка жінок з переломом (%)+ |
Зниження абсолютного ризику (%) (95 % ДІ) |
Зниження відносного ризику (%) (95 % ДІ) |
|
|
Плацебо, n =3D 3906 |
Проліа®, n =3D 3902 |
|||
|
Будь-який клінічний перелом1 |
10,2 |
7,2 |
2,9 (1,6, 4,2) |
30 (19, 41)*** |
|
Клінічний вертебральний перелом |
2,6 |
0,8 |
1,8 (1,2, 2,4) |
69 (53, 80)*** |
|
Невертебральний перелом2 |
8,0 |
6,5 |
1,5 (0,3, 2,7) |
20 (5, 33)** |
|
Головний невертебральний перелом3 |
6,4 |
5,2 |
1,2 (0,1, 2,2) |
20 (3, 34)* |
|
Головний остеопоротичний перелом4 |
8,0 |
5,3 |
2,7 (1,6, 3,9) |
35 (22, 45)*** |
*p ≤ 0,05; **p =3D 0,0106 (вторинна кінцева точка включена в регулювання кратності), ***р ≤ 0,0001
+ Частота подій базується на розрахунку згідно з методом Каплана-Мейєра протягом 3 років.
1Включає клінічні вертебральні та невертебральні переломи.
2Виключає переломи хребців, черепа, обличчя, нижньої щелепи, п'ясті і фаланг пальців рук і ніг.
3Включає таз, дистальний відділ стегна, проксимальний відділ великогомілкової кістки, ребра, проксимальний відділ плечової кістки, передпліччя і стегно.
4Включає клінічні вертебральні переломи, переломи шийки стегна, передпліччя і плечової кістки, як це визначено ВООЗ.
У жінок з вихідним рівнем МЩК шийки стегна ≤ -2,5 препарат Проліа® знижує ризик невертебральних переломів (35 % зниження відносного ризику, 4,1 % зниження абсолютного ризику, р < 0,001, розвідувальний аналіз).
Зниження частоти виникнення нових вертебральних переломів, переломів шийки стегна і невертебральних переломів через застосування Проліа® протягом 3 років були послідовними незалежно від 10-річного вихідного ризику переломів.
Вплив на мінеральну щільність кісток
Порівняно з плацебо на 1, 2 і 3 році препарат Проліа® значно підвищив МЩК: на 9,2 % в поперековому відділі хребта, на 6,0 % в тазостегновому зчленуванні, на 4,8 % в шийці стегнової кістки, на 7,9 % у вертлюзі стегнової кістки, на 3,5 % у дистальному 1/3 відділі променевої кістки і на 4,1 % у всьому організмі протягом 3 років (всі р < 0,0001).
У клінічних дослідженнях ефектів припинення застосування препарату Проліа® МЩК повернулась приблизно до рівнів, які спостерігались перед лікуванням, і залишалася вищою, ніж у групі плацебо, протягом 18 місяців після останньої дози. Ці дані вказують на те, що подальше лікування препаратом Проліа® необхідне для підтримання ефекту лікарського засобу. Повторне застосування препарату Проліа® призвело до підвищення МЩК, подібного до такого як при першому застосуванні препарату Проліа®.
Відкрите розширене дослідження лікування постменопаузального остеопорозу
В цілому 4550 жінок (2343 з яких застосовували Проліа® і 2207 - плацебо), які пропустили не більше однієї дози препарату в базовому дослідженні, описаному вище, і завершили візит, передбачений дослідженням на 36 місяці, брали участь в 7-річному багатонаціональному багатоцентровому відкритому непорівняльному розширеному дослідженні довгострокової безпеки і ефективності препарату Проліа®. Всі жінки в розширеному дослідженні повинні були отримувати 60 мг Проліа® кожні 6 місяців, а також щоденно кальцій (принаймні 1 г) і вітамін D (принаймні 400 МО). В цілому 2626 пацієнток (58 % жінок, включених в розширене дослідження, тобто 34 % жінок, включених в базове дослідженні) завершили розширене дослідження.
У пацієнтів, які отримували Проліа® протягом періоду тривалістю до 10 років, МЩК збільшилась від вихідних даних основного дослідження на 21,7 % в поперековому відділі хребта, на 9,2 % в тазостегновому зчленуванні, на 9,0 % в шийці стегнової кістки, 13,0 % у вертлюзі і на 2,8 % в дистальному 1/3 відділі променевої кістки. Наприкінці дослідження у пацієнтів, які лікувалися протягом 10 років, середній Т-показник МЩК поперекового відділу хребта становив -1,3.
Частота виникнення переломів оцінювалась як кінцева точка безпеки, однак ефективність запобігання переломам оцінити не можна внаслідок великої кількості випадків припинення лікування та відкритого формату дослідження. У пацієнтів, які приймали деносумаб протягом 10 років (n =3D 1278), сумарна кількість нових випадків вертебральних та невертебральних переломів становила приблизно 6,8 % та 13,1 % відповідно. У пацієнтів, які з будь-яких причин не завершили дослідження, частота виникнення переломів протягом періоду лікування була більш високою.
Тринадцять встановлених випадків остеонекрозу щелепи (ОНЩ) і два встановлені випадки атипових переломів стегнової кістки спостерігались в ході розширеного дослідження.
Лікування остеопорозу у чоловіків
Ефективність та безпеку препарату Проліа®, який застосовували 1 раз на 6 місяців протягом 1 року, досліджували у 242 чоловіків у віці 31-84 років. Суб'єкти з рШКФ < 30 мл/хв/1,73 м2 були виключені з дослідження. Всі чоловіки отримували добавки кальцію (принаймні 1000 мг) і вітаміну D (принаймні 800 МО) щодня.
Основним показником ефективності була відсоткова зміна показника МЩК поперекового відділу хребта, ефективність щодо переломів не оцінювали. Порівняно з плацебо препарат Проліа® значно збільшив МЩК протягом 12 місяців: на 4,8 % в поперековому відділі хребта, на 2,0 % в тазостегновому зчленуванні, на 2,2 % в шийці стегнової кістки, на 2,3 % у вертлюзі стегнової кістки і на 0,9 % у дистальному 1/3 відділі променевої кістки (всі р < 0,05). Показник МЩК поперекового відділу хребта збільшився від вихідних даних у 94,7 % чоловіків на кінець 1 року. Значне збільшення МЩК в поперековому відділі хребта, тазостегновому зчленуванні, шийці стегнової кістки і вертлюзі стегнової кістки спостерігалося через 6 місяців (р < 0,0001).
Гістологія кісток
Гістологію кісток оцінювали у 62 жінок в постменопаузальний період з остеопорозом або низькою кістковою масою, які раніше не отримували терапії з приводу остеопорозу або які перейшли з попередньої терапії алендронатом та після 1- 3 років лікування із застосуванням Проліа®. П'ятдесят дев'ять жінок взяли участь в піддослідженні кісткової біопсії на 24 місяці (n =3D 41) і/або 84 місяці (n =3D 22) розширеного дослідження, проведеного за участю жінок в постменопаузальному періоді з остеопорозом. Гістологію кісток також оцінювали у 17 чоловіків з остеопорозом після 1 року лікування препаратом Проліа®. Результати біопсії кісток показали кістку нормальної форми та якості без ознак дефектів мінералізації, незрілої кістки або фіброзу кісткового мозку. Результати гістоморфометрії в розширеному дослідженні показали, що у жінок в постменопаузальний період з остеопорозом антирезорбтивні ефекти Проліа®, які вимірювались за частотою активації і швидкості формування кісток, підтримувались протягом тривалого часу.
Лікування втрати кісткової тканини, пов'язаної з андрогенною депривацією
Ефективність та безпеку препарату Проліа®, який застосовували 1 раз на 6 місяців протягом 3 років, досліджували у чоловіків з гістологічно підтвердженим неметастатичним раком передміхурової залози, які отримували андроген деприваційну терапію (1468 чоловіків у віці 48-97 років). Ці пацієнти були схильні до підвищеного ризику перелому (визначається як вік > 70 років або вік < 70 років і Т-показник МЩК в поперековому відділі хребта, тазостегновому зчленуванні або шийці стегнової кістки < -1,0 або наявність остеопоротичного перелому в анамнезі ). Всі чоловіки отримували добавки кальцію (принаймні 1000 мг) і вітаміну D (принаймні 400 МО) щодня.
Порівняно з плацебо препарат Проліа® значно підвищив МЩК протягом 3 років: на 7,9 % в поперековому відділі хребта, на 5,7 % в тазостегновому зчленуванні, на 4,9 % в шийці стегнової кістки, на 6,9 % в вертлюзі стегнової кістки, на 6,9 % у дистальному 1/3 відділі променевої кістки і на 4,7 % у всьому організмі (всі р < 0,0001). У проспективно запланованому розвідувальному аналізі спостерігалося значне збільшення МЩК в поперековому відділі хребта, тазостегновому зчленуванні, шийці стегнової кістки і вертлюзі стегнової кістки через 1 місяць після застосування початкової дози.
Препарат Проліа® продемонстрував значне зниження відносного ризику нових переломів хребців: 85 % (1,6 % зниження абсолютного ризику) через 1 рік, 69 % (2,2 % зниження абсолютного ризику) через 2 роки і 62 % (2,4 % зниження абсолютного ризику) через 3 роки (всі р < 0,01).
Лікування втрати кісткової маси, пов'язаної з ад'ювантною терапією інгібітором ароматази
Ефективність та безпеку препарату Проліа®, який застосовували 1 раз кожні 6 місяців протягом 2 років, досліджували у жінок з неметастатичним раком молочної залози (252 жінки у віці 35-84 років) і з базовим рівнем Т-показника МЩК між -1,0 та -2,5 в поперековому відділі хребта, тазостегновому зчленуванні або шийці стегнової кістки. Всі жінки отримували добавки кальцію (принаймні 1000 мг) і вітаміну D (принаймні 400 МО) щодня.
Основним показником ефективності була відсоткова зміна показника МЩК поперекового відділу хребта, ефективність щодо переломів не оцінювали. Порівняно з плацебо препарат Проліа® значно збільшив МЩК протягом 2 років: на 7,6 % в поперековому відділі хребта, 4,7 % в тазостегновому зчленуванні, на 3,6 % в шийці стегнової кістки, на 5,9 % у вертлюзі стегнової кістки, на 6,1 % в дистальному 1/3 відділі променевої кістки і на 4,2 % у всьому організмі (всі р < 0,0001).
Дитяча популяція
Європейське агентство з лікарських засобів скасувало зобов'язання надавати результати досліджень препарату Проліа® у всіх підмножинах педіатричної популяції при лікуванні втрати кісткової маси, пов'язаної з аблятивною терапією статевими гормонами, і у підмножинах педіатричної популяції у віці до 2 років при лікуванні остеопорозу (див. розділ «Спосіб застосування та дози» для отримання інформації щодо застосування в педіатрії).
Фармакокінетика.
Абсорбція
Після підшкірного введення у дозі 1,0 мг/кг, що приблизно відповідає дозі 60 мг з огляду на показники AUC, експозиція становила 78 % від рівня, отриманого при внутрішньовенному введенні цієї ж дози препарату. Після підшкірного введення 60 мг деносумабу максимальна сироваткова концентрація деносумабу (Cmax), що становить в середньому 6 мкг/мл (діапазон - 1-17 мкг/мл), досягається в середньому за 10 днів (діапазон - 2-28 днів).
Біотрансформація
Деносумаб складається виключно з амінокислот та вуглеводів, як і природний імуноглобулін. Тому малоймовірно, що він виводиться шляхом печінкового метаболізму. Вважається, що його метаболізм та виведення відбуваються тими ж шляхами, що і кліренс імуноглобуліну, після розпаду препарату на невеликі пептиди та окремі амінокислоти.
Виведення
Після досягнення Cmax сироватковий рівень препарату знижується протягом 3 місяців (діапазон - 1,5-4,5 місяця) у зв'язку з періодом напіввиведення, що становить 26 днів (діапазон - 6-52 дні). У 53 % пацієнтів через 6 місяців після застосування препарату деносумаб не виявлявся.
При багаторазовому застосуванні деносумабу в режимі 60 мг підшкірно 1 раз кожні 6 місяців не спостерігалося ані кумуляції препарату, ані змін його фармакокінетики з часом. На фармакокінетику деносумабу не впливало формування зв'язків антитіл з деносумабом, та фармакокінетика препарату була однаковою у жінок і чоловіків. Вік (28-87 років), расова приналежність, стан хвороби (втрата кісткової маси або остеопороз, рак передміхурової залози або рак молочної залози) не мали суттєвого впливу на фармакокінетику деносумабу.
Спостерігалася тенденція до збільшення маси тіла та зменшення експозиції препарату з огляду на показники AUC та Cmax. Однак така тенденція не вважається клінічно значущою, оскільки фармакодинамічний ефект оцінюється за маркерами ремоделювання кісток та збільшенням мінеральної щільності кісток, що були постійними у різних вагових категоріях хворих.
Лінійність/нелінійність
У ході досліджень дозозалежності був виявлений нелінійний зв'язок фармакокінетики препарату з його дозою зі зменшенням кліренсу препарату при збільшенні його дози, але приблизне пропорційне дозозалежне зростання експозиції препарату спостерігається при застосуванні доз від 60 мг.
Дані доклінічних досліджень
В дослідженнях токсичності однократної та багатократних доз на яванських макаках дози деносумабу, які призводили до системної відповіді, вищі в 100-150 разів відрекомендованих для людини, не впливали на фізіологію серцево-судинної системи, репродуктивну функцію самців або самок або на виникнення специфічної токсичності для органів-мішеней.
В дослідженні у макак, яким вводили деносумаб протягом І триместру вагітності, при дозуванні на рівні AUC, до 99 разів вищому за дозування у людини (60 мг кожні 6 місяців), не було виявлено негативного впливу на організми матері та плода. В цьому досліджені лімфатичні вузли плода не вивчали.
В іншому дослідженні у макак, яким вводили деносумаб протягом вагітності при дозуванні на рівні AUC, у 119 разів вищому за дозування у людини (60 мг кожні 6 місяців), було виявлено: підвищення рівнів мертвонародження та постнатальної летальності; порушення росту кісток, які проявлялися зниженням міцності кісток, зниженням гематопоезу та затримкою прорізування зубів; відсутність периферичних лімфатичних вузлів та зниження неонатального росту плода. Розвиток молочних залоз макак не відрізнявся від норми.
У доклінічних дослідженнях, проведених на нокаутних мишах, позбавлених RANK або RANKL, спостерігалося погіршення формування лімфатичних вузлів у плода. Відсутність лактації за рахунок пригнічення дозрівання молочної залози (дольчато-альвеолярний розвиток залози під час вагітності) спостерігалась також в нокаутних мишей, позбавлених RANK або RANKL.
Спеціальні групи хворих
Ниркова недостатність
У дослідженні за участю 55 пацієнтів з різними стадіями ниркової недостатності, включаючи пацієнтів, які потребували діалізу, рівень ниркової недостатності не впливав на фармакокінетику деносумабу.
Печінкова недостатність
Спеціальних досліджень за участю пацієнтів з печінковою недостатністю не проводилось. Загалом моноклональні антитіла не виводяться шляхом печінкового метаболізму, тому очікується, що печінкова недостатність не буде мати впливу на фармакокінетику деносумабу.
Дитяча популяція
Фармакокінетичний профіль у педіатричній популяції не оцінювався.
Клінічні характеристики
Показання
Лікування остеопорозу у жінок у постменопаузальному періоді та у чоловіків із підвищеним ризиком переломів. У жінок у постменопаузальному періоді препарат Проліа® значно зменшує ризик переломів хребців, переломів нехребцевої локалізації та переломів стегна.
Лікування втрати кісткової маси у чоловіків з підвищеним ризиком переломів хребців, які отримують гормоносупресивну терапію у зв'язку з раком передміхурової залози.
У чоловіків з раком передміхурової залози, які отримують гормоносупресивну терапію, Проліа® значно знижує ризик переломів хребців.
Протипоказання
- Гіперчутливість до діючої речовини або до будь-якого з допоміжних компонентів препарату;
- Гіпокальціємія (див. розділ «Особливості застосування»).
Взаємодія з іншими лікарськими засобами та інші види взаємодій
Клінічних даних стосовно взаємодії деносумабу та гормональної замісної терапії (естрогенів) немає, однак потенційна можливість фармакодинамічної взаємодії вважається низькою.
За даними дослідження, у жінок у постменопаузальному періоді, які були переведені з попередньої терапії алендронатом на лікування деносумабом, фармакокінетика та фармакодинаміка деносумабу не змінилися після попереднього застосування алендронату.
У дослідженні взаємодії було встановлено, що препарат Проліа® (60 мг підшкірно) не впливає на фармакокінетику мідазоламу, що метаболізується за допомогою цитохрому Р450 3А4 (CYP3A4). Це свідчить про відсутність впливу Проліа® на фармакокінетику лікарських засобів, що метаболізуються цим ферментом.
Особливості застосування
Поповнення кальцію та вітаміну D
Для всіх пацієнтів дуже важливим є адекватне вживання кальцію та вітаміну D.
Застережні заходи при застосуванні
Гіпокальціємія
Важливо ідентифікувати пацієнтів з ризиком виникнення гіпокальціємії та скорегувати гіпокальціємію за допомогою адекватного вживання кальцію та вітаміну D до початку лікування препаратом. Рекомендований клінічний моніторинг рівнів кальцію у крові пацієнтів, схильних до розвитку гіпокальціємії, в перші два тижні після початкової дози. Якщо у будь-якого пацієнта на тлі лікування препаратом можна запідозрити симптоми гіпокальціємії (див. розділ «Побічні реакції»), слід визначити рівень кальцію. Пацієнтам слід рекомендувати повідомляти про симптоми, які вказують на гіпокальціємію.
В умовах постмаркетингового спостереження були отримані повідомлення про важку симптоматичну гіпокальціємію (див. розділ «Побічні реакції»), яка в більшості випадків виникала в перші тижні після початку лікування, але може також виникати пізніше.
Інфекції шкіри
У пацієнтів, які приймають препарат Проліа®, можуть виникнути інфекції шкіри (переважно целюліт), що призводять до госпіталізації (див. розділ «Побічні реакції»). Пацієнтам необхідно негайно звернутися за медичною допомогою, якщо у них з'являться симптоми целюліту.
Остеонекроз щелепи
Отримані рідкісні повідомлення про остеонекроз щелепи у пацієнтів, які перебувають на лікуванні остеопорозу препаратом Проліа® (див. розділ «Побічні реакції»).
Пацієнтам з ураженнями м'яких тканин ротової порожнини, які не загоюються, слід на один місяць відкласти початок/новий курс лікування.
Перед лікуванням препаратом Проліа® хворим із супутніми факторами ризику потрібна попередня консультація стоматолога з проведенням відповідних профілактичних заходів та індивідуальної оцінки співвідношення ризику/користі.
Фактори, які слід взяти до уваги при оцінці ризику розвитку у пацієнта остеонекрозу щелепи:
- сила лікарського засобу, який гальмує резорбцію кістки (більший ризик для більш потужних сполук), шлях введення (більший ризик для парентерального введення) та кумулятивна доза препарату для лікування резорбції кістки;
- рак, супутні стани (наприклад анемія, коагулопатії, інфекції), паління;
- супутня терапія: кортикостероїди, хіміотерапія, інгібітори ангіогенезу, радіотерапія голови та шиї;
- погана гігієна порожнини рота, захворювання періодонту, невідповідне протезування зубів, хвороба зубів в анамнезі, інвазивні стоматологічні процедури, наприклад екстракції зубів.
Всі пацієнти повинні дотримуватися відповідних правил гігієни порожнини рота, проходити регулярні профілактичні огляди у стоматолога та негайно повідомляти про будь-які симптоми з боку ротової порожнини, включаючи рухливість зубів, біль або набряки, рани, які не загоюються, або виділення з ран протягом лікування препаратом Проліа®.
Якщо після уважного розгляду прийнято рішення щодо необхідності проведення інвазивних стоматологічних процедур, їх не слід проводити у дні безпосередньо перед або відразу після введення препарату Проліа®.
виникнення остеонекрозу щелепи під час лікування препаратом потребує проведення лікарем пацієнта разом із стоматологом або хірургом клінічної оцінки та складання плану лікування пацієнта, що базується на індивідуальній оцінці співвідношення ризику/користі з тимчасовою зупинкою лікування препаратом Проліа® поки остеонекроз щелепи не буде подолано та фактори ризику не будуть пом'якшені.
Остеонекроз зовнішнього слухового каналу
Повідомлялося про випадки остеонекрозу зовнішнього слухового каналу, що виникали при лікуванні деносумабом. До можливих чинників ризику виникнення остеонекрозу зовнішнього слухового каналу належать застосування стероїдів і хіміотерапія та/або локальні чинники ризику, такі як інфекція або травма. Вірогідність виникнення остеонекрозу зовнішнього слухового каналу необхідно враховувати у разі лікування деносумабом пацієнтів з порушеннями з боку вуха, включаючи хронічні інфекції вуха.
Атипові переломи стегна
У хворих, які лікувались препаратом Проліа®, повідомлялось про випадки атипових переломів стегна (див. розділ «Побічні реакції»). Атипові переломи стегна можуть виникати при незначних травмах або при відсутності травм у підвертлюжній або діафізальній ділянці стегна та можуть бути двобічними. Ці переломи характеризуються специфічними радіографічними показниками. Про атипові переломи стегна також повідомлялось у пацієнтів з певними коморбідними станами (наприклад дефіцитом вітаміну D, ревматоїдним артритом, гіпофосфатазією) та при застосуванні певних фармацевтичних засобів (наприклад, біфосфонатів, глюкокортикоїдів, інгібіторів протонної помпи). Такі явища також виникали без антирезорбтивної терапії. Подібні переломи, про які повідомлялося у зв'язку з прийомом біфосфонатів, часто є білатеральними; таким чином, протягом терапії препаратом Проліа® необхідно обстежувати протилежне стегно у пацієнтів з підтвердженим переломом тіла стегнової кістки.
Потрібно розглянути відміну препарату Проліа® для пацієнтів з підозрою на атиповий перелом стегна під час оцінки пацієнта на основі індивідуального співвідношення користь/ризик.
Під час лікування препаратом Проліа® пацієнтів слід попередити про необхідність повідомляти про нові або незвичайні випадки болю у стегновій кістці, стегні або у паховій ділянці. Пацієнтів, які мають такі симптоми, необхідно обстежити на наявність неповних переломів стегна.
Тривале лікування препаратами антирезорбтивної дії
Тривале лікування препаратами антирезорбтивної дії (включаючи як деносумаб, так і біфосфонати) може призводити до підвищення ризику виникнення побічних ефектів, таких як остеонекроз щелепи й атипові переломи стегна, внаслідок суттєвого пригнічення процесу ремоделювання кісткової тканини (див. розділ «Спосіб застосування та дози»).
Сухий природний каучук
Ковпачок голки на попередньо заповненому шприці одноразового використання містить сухий природний каучук (похідна латексу), що може спричинити алергічну реакцію.
Сумісне лікування з іншими лікарськими засобами, що містять деносумаб
Пацієнти, які лікуються препаратом Проліа®, не повинні одночасно приймати інші лікарські засоби, що містять деносумаб (для профілактики уражень кісткової системи у дорослих хворих з метастазами у кістки із солідних новоутворень).
Ниркова недостатність
У пацієнтів з тяжкою нирковою недостатністю (кліренс креатиніну < 30 мл/хв) або у тих, хто перебуває на діалізі, збільшується ризик розвитку гіпокальціємії. Ризик розвитку гіпокальціємії та супутнього підвищення рівнів паратгормону підвищується зі збільшенням ступеня ниркової недостатності. Для цих пацієнтів особливо важливі адекватне вживання кальцію, вітаміну D та регулярний моніторинг рівня кальцію (див. початок розділу).
Застереження щодо допоміжних речовин
Цей лікарський засіб містить сорбіт.
Пацієнти із рідкісною вродженою непереносимістю фруктози не повинні застосовувати препарат Проліа®.
Препарат містить менше ніж 1 ммоль натрію (23 мг) на 60 мг, тобто, по суті, є вільним від натрію.
Застосування у період вагітності або годування груддю.
Вагітність
Дані про безпеку застосування препарату Проліа® вагітним наразі відсутні. В дослідженні на макаках, яким вводили деносумаб протягом вагітності при дозуванні на рівні AUC у 119 разів вищому за дозування у людини, було виявлено репродуктивну токсичність (див. розділ «Фармакологічні властивості»).
Препарат Проліа® не рекомендований для застосування у період вагітності.
Годування груддю
Невідомо, чи екскретується деносумаб у грудне молоко людини. Дослідження на нокаутних мишах (генетично модифіковані миші, у яких RANKL, що є мішенню для деносумабу, відключено через видалення гена, (див. розділ «Фармакологічні властивості»)) дають змогу припустити, що відсутність RANKL впливає на дозрівання молочних залоз, що призводить до порушень годування груддю після пологів (див. розділ «Фармакологічні властивості»). Рішення про відмову від грудного вигодовування або від лікування препаратом Проліа® слід приймати, зважуючи переваги грудного вигодовування для немовляти та лікування препаратом Проліа® для матері.
Фертильність
Даних про вплив деносумабу на фертильність людини немає. Дослідження на тваринах не вказують на наявність прямих або непрямих негативних ефектів застосування препарату Проліа® на фертильність (див. розділ «Фармакологічні властивості»).
Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами.
Препарат Проліа® не має або має дуже незначний вплив на швидкість реакції при керуванні автотранспортом або роботі з іншими механізмами.
Спосіб застосування та дози
Препарат застосовують підшкірно.
Дозування
Рекомендована доза препарату Проліа® - одна підшкірна ін'єкція 60 мг препарату 1 раз кожні 6 місяців, що вводиться у стегно, живіт або зовнішню поверхню плеча.
Протягом терапії пацієнти як доповнення до лікування повинні отримувати препарати та харчові добавки, що містять кальцій та вітамін D (див. розділ «Особливості застосування»).
Оптимальна загальна тривалість лікування остеопорозу шляхом застосування препаратів антирезорбтивної дії (включаючи як деносумаб, так і біфосфонати) не була визначена. Необхідно періодично переглядати питання щодо потреби у безперервному лікуванні, враховуючи користь і потенційний ризик від застосування деносумабу для кожного окремого пацієнта, особливо через 5 або більше років лікування (див. розділ «Особливості застосування»).
Спосіб застосування
Препарат для підшкірного введення.
Препарат повинні застосовувати пацієнти, які пройшли інструктаж з техніки ін'єкційного введення.
Особливі запобіжні заходи при поводженні з препаратом і його утилізації
Перед застосуванням розчин Проліа® необхідно оглянути на наявність твердих часточок або зміни кольору. Розчин не можна використовувати, якщо він містить часточки чи помутнів або змінив свій колір. Не струшувати. Для запобігання дискомфорту у місці ін'єкції необхідно дати попередньо заповненому шприцу нагрітися до кімнатної температури (до 25 °C) перед ін'єкцією та вводити препарат повільно. Потрібно ввести весь вміст попередньо заповненого шприца. Будь-яку кількість лікарського засобу, що залишилася у попередньо заповненому шприці після ін'єкції, слід знищити згідно з діючими вимогами.
Ниркова недостатність
Змінювати дозу препарату для лікування хворих з нирковою недостатністю не потрібно (див. розділ «Особливості застосування» щодо рекомендацій стосовно контролю за вмістом кальцію).
Печінкова недостатність
Безпека та ефективність деносумабу для лікування пацієнтів з печінковою недостатністю не вивчалися (див. розділ «Фармакокінетика»).
Хворі літнього віку (віком ≥ 65 років)
Змінювати дозу препарату для лікування пацієнтів літнього віку не потрібно.
Діти
Препарат Проліа® не рекомендований для застосування дітям (вік < 18 років), оскільки його безпека та ефективність для пацієнтів дитячого віку не встановлені. В експериментальних дослідженнях на тваринах інгібування рецептора-активатора ядерного фактора kB (RANK)/ліганду RANK (RANKL) супроводжувалося інгібуванням росту кісток та затримкою прорізування зубів.
Вказівки для пацієнта щодо проведення ін'єкції за допомогою попередньо заповненого шприца з голкою, закритою ковпачком, із захисним пристроєм від випадкового уколу голкою
|
Схема пристрою |
|||||||||||||||||
|
До застосування |
Після застосування |
||||||||||||||||
|
|
|
|
||||||||||||||
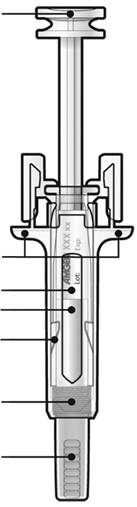
Перед застосуванням попередньо заповненого шприца з голкою, закритою ковпачком, із захисним пристроєм проти випадкового уколу голкою, прочитайте цю важливу інформацію:
- Не виконуйте ін'єкції самостійно до отримання інструкцій від Вашого лікаря чи медичного працівника.
- Препарат Проліа® вводять шляхом ін'єкції у тканину, що розташована зразу під шкірою (підшкірна ін'єкція).
- Повідомте лікаря, якщо у вас є алергія на латекс (ковпачок для голки на попередньо заповненому шприці містить похідну латексу).
- Не видаляйте сірий ковпачок для голки з попередньо заповненого шприца поки Ви не будете готові до ін'єкції.
- Не використовуйте попередньо заповнений шприц, якщо перед тим він впав на тверду поверхню. Застосовуйте новий попередньо заповнений шприц та зверніться до свого лікаря або медичного працівника.
- Не активуйте попередньо заповнений шприц, якщо Ви не збираєтесь виконувати ін'єкцію.
- Не знімайте прозорий захисний пристрій від випадкового уколу голкою з попередньо заповненого шприца.
У разі виникнення питань зверніться до свого лікаря або медичного працівника.
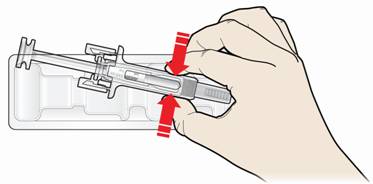
Крок 1: Підготовка.
А. Витягніть контейнер з попередньо заповненим шприцом з упаковки та підготуйте необхідні для ін'єкції приладдя.
Для комфортної ін'єкції витримайте попередньо заповнений шприц при кімнатній температурі протягом 30 хвилин перед ін'єкцією. Ретельно вимийте свої руки з милом.
Покладіть шприц на добре освітлений чистий письмовий стіл. Знайдіть спиртові серветки, ватний тампон, марлеву серветку або пластир, а також контейнер для утилізації гострих відходів (не входять в комплект).
- Не намагайтеся зігріти шприц за допомогою джерела тепла, наприклад гарячою водою або у мікрохвильовій печі.
- Не залишайте попередньо заповнений шприц під прямими сонячними променями.
- Не струшуйте попередньо заповнений шприц.
- Зберігайте попередньо заповнені шприці у місцях, недоступних для дітей та захищених від світла.
Б. Відкрийте контейнер шприца, видаливши покриття. Видаліть попередньо заповнений шприц з контейнера.
|
|
Для безпечного застосування: - Не торкайтеся поршня. - Не торкайтеся сірого ковпачка для голки. |
|
Тримати тут |
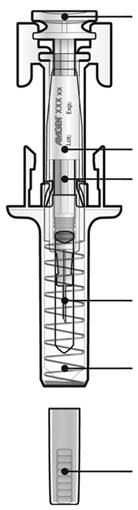
В. Проведіть огляд лікарського засобу та попередньо заповненого шприца.
Не використовуйте попередньо заповнений шприц, якщо:
- Лікарський засіб непрозорий або має тверді часточки, розчин має бути прозорим, безбарвним або жовтуватого кольору.
- Будь-яка частина шприца має тріщини або зламана.
- Сірий ковпачок для голки відсутній або голка ним не повністю прикрита.
- Сплинув останній день місяця, який вказано на етикетці як кінцевий термін використання.
У всіх зазначених випадках зверніться до свого лікаря або медичного працівника.
Крок 2: Готовність.
А. Ретельно вимийте руки. Підготуйте та очистіть місце ін'єкції.
|
Місця для ін'єкції |
|
|
|
Зовнішня поверхня плеча (тільки якщо хтось проводить Вам ін'єкцію) Живіт, окрім 5-сантиметрової зони довкола пупка Верхня частина стегна |
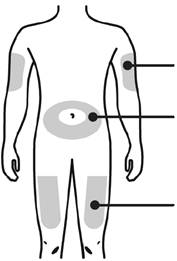
Обробіть місце ін'єкції за допомогою серветки з етиловим спиртом. Дочекайтесь, щоб шкіра стала сухою.
- Не торкайтеся місця ін'єкції до її проведення.
- Не виконуйте ін'єкції у місця, де шкіра тонка, незвичайного кольору, напружена або порушена її цілісність. Уникайте ін'єкції в місця шрамів і розтяжок.
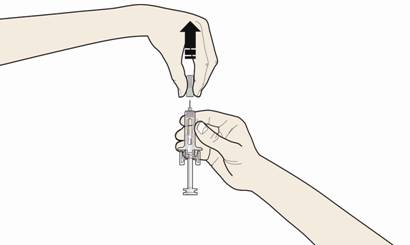
Б. Обережно зніміть сірий ковпачок з голки у напрямку, протилежному від вашого тіла.
В. Затисніть місце ін'єкції, щоб створити міцну поверхню.
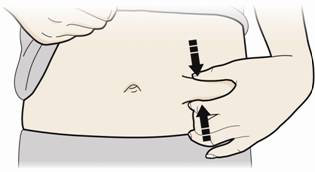
- Важливо тримати шкіру затиснутою при введенні.
Крок 3: Ін'єкція.
А. Тримаючи шкіру затиснутою, ВВЕДІТЬ голку у шкіру.
- Не торкайтеся обробленої ділянки шкіри.
Б. Повільно та постійно тисніть на поршень, просуваючи його, доки не відчуєте або не почуєте звук «тріску». Дотисніть поршень донизу.
|
|
||
|
|
"ТРІСК" |
|



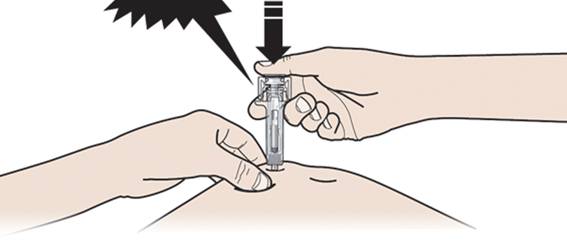
Важливо натискувати на поршень до кінця до появи тріску задля введення повної дози.
В. Відпустіть великий палець. Потім підніміть шприц над шкірою.
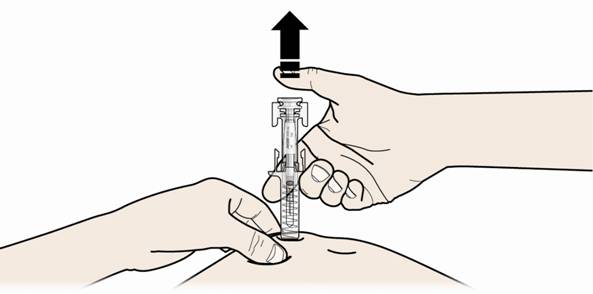
Після того, як Ви відпустили поршень, захисний пристрій від випадкового уколу голкою накриє голку.
- Не повертайте ковпачок на шприц, що був попередньо заповненим.
Крок 4: Завершення.
А. Утилізуйте використаний шприц та інші матеріали у контейнер для утилізації гострих відходів.
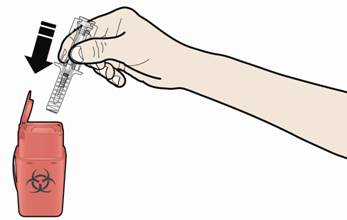
Ліки мають бути утилізовані відповідно до місцевих вимог. Не допускайте їх потрапляння в навколишнє середовище.
Зберігати шприц та контейнер для гострих відходів в недоступному для дітей місці та захищеному від світла місці.
- Повторно не використовуйте шприц, що був попередньо заповнений.
- Не переробляйте попередньо заповнені шприци та не викидайте їх у побутові відходи.
Б. Проведіть огляд місця ін'єкції.
Якщо є кров, притисніть ватний або марлевий тампон до місця ін'єкції. Не тріть місце ін'єкції. Нанесіть пластир, якщо це необхідно.
При утилізації використаних шприців:
• НЕ встановлюйте ковпачок для голки на використаний шприц.
• Тримайте використані шприци в недоступному для дітей місці.
Використані шприци потрібно утилізувати відповідно до місцевих вимог. Не допускайте їх потрапляння в навколишнє середовище.
Передозування.
Не було отримано даних стосовно передозування препарату. При дозах до 180 мг кожні 4 тижні (кумулятивна доза - до 1080 мг за 6 місяців) не спостерігалося інших, окрім уже наведених, побічних ефектів.
Побічні реакції
Резюме профілю безпеки
Загальний профіль безпеки препарату Проліа® був однаковим у пацієнтів з остеопорозом та у пацієнтів з раком молочної залози або раком простати, які отримують гормоносупресивну терапію у п'ятьох плацебо-контрольованих клінічних дослідженнях III фази.
Найбільш поширені побічні ефекти при застосуванні препарату Проліа® (які спостерігались в більш ніж одного пацієнта з десяти) є скелетно-м'язовий біль і біль в кінцівках. Нечасті випадки целюліту; рідкісні випадки гіпокальціємії, гіперчутливості, остеонекрозу щелепи і атипових переломів стегнової кістки (див. розділ «Особливості застосування» і розділ «Побічні реакції»: Опис окремих побічних реакцій) спостерігалися у пацієнтів, які приймають препарат Проліа®.
Табличний список побічних реакцій
В таблиці 3 описані побічні реакції, щодо яких були повідомлення в ході клінічних випробувань фази II і III при застосуванні у хворих з остеопорозом і раком молочної залози або раком простати, які отримують гормональну терапію і/або спонтанні повідомлення.
Побічні реакції класифіковані у такі групи відповідно до частоти їх виникнення (див. табл. 3):
дуже часто(≥ 1/10), часто(≥ 1/100 та < 1/10), нечасто(≥ 1/1000 та < 1/100), рідко (≥ 1/10000 та < 1/1000), дуже рідко (< 1/10000) та невідома.
У межах кожної групи побічні реакції вказані за частотою та системою та представлені в порядку зменшення частоти виникнення.
Таблиця 3. Побічні реакції у пацієнтів з остеопорозом і раком молочної залози або раком простати, які отримують гормоносупресивну терапію
|
Клас системи органів за MedDRA |
Категорія частоти |
Побічний ефект |
|
Інфекційні та паразитарні захворювання |
Часто Часто Нечасто Нечасто Нечасто |
Інфекції сечовидільної системи Інфекції верхніх відділів дихальної системи Дивертикуліт1 Целюліт1 Інфекції вуха |
|
Розлади з боку імунної системи |
Рідко Рідко |
Медикаментозна гіперчутливість1 Анафілактична реакція1 |
|
Метаболічні та аліментарні розлади |
Рідко |
Гіпокальціємія1 |
|
Розлади з боку нервової системи |
Часто |
Ішіас |
|
Розлади з боку шлунково-кишкового тракту |
Часто Часто |
Запор Дискомфорт у животі |
|
Розлади з боку шкіри та підшкірної клітковини |
Часто Часто |
Висипання Екзема |
|
Розлади з боку опорно-рухового апарату та сполучної тканини |
Дуже часто Дуже часто |
Біль у кінцівках Біль у м'язах та кістках1 |
|
Рідко Рідко Невідома |
Остеонекроз щелепи1 Атипові переломи стегна1 Остеонекроз зовнішнього слухового каналу2 |
1 Див. розділ «Опис окремих побічних реакцій».
2 Див. розділ «Особливості застосування».
В узагальненому аналізі даних усіх плацебо-контрольованих досліджень II і III фази повідомлялося про захворюваність грипом. Яка становила 1,2 % для деносумабу і 0,7 % для плацебо. Однак, цей дисбаланс був встановлений в узагальненому аналізі даних і не виявлений при стратифікованому аналізі.
Опис окремих побічних реакцій
Гіпокальціємія
У двох плацебо-контрольованих клінічних дослідженнях III фази у жінок в постменопаузі з остеопорозом, приблизно 0,05 % (2 з 4050) мали зниження сироваткових рівнів кальцію (менше 1,88 ммоль/л) після введення препарату Проліа®. Зниження сироваткових рівнів кальцію (менше 1,88 ммоль/л) не повідомлялось ні в двох плацебо-контрольованих клінічних дослідженях фази III у пацієнтів, які отримують гормональну терапію, ні в плацебо‑контрольованому клінічному дослідженні фази III у чоловіків з остеопорозом.
За даними постмаркетингового спостереження, повідомлялися рідкісні випадки тяжкої симптоматичної гіпокальціємії, переважно у пацієнтів з підвищеним ризиком виникнення гіпокальціємії, які приймали препарат Проліа®, та переважно у перші тижні після початку терапії. Випадки клінічних проявів тяжкої симптоматичної гіпокальціємії включали пролонгацію інтервалу QT, тетанію, судоми і зміни психічного стану (див. розділ «Особливості застосування»). Симптоми гіпокальціємїї, які було виявлено у ході клінічних досліджень із застосуванням деносумабу, включали парестезії або жорсткість м'язів, посмикування, спазми і м'язові cудоми.
Шкірні інфекції
У плацебо-контрольованих клінічних дослідженнях фази III, загальна частота інфекцій шкіри була подібною у групі плацебо і групі, де приймали препарат Проліа®: у жінок в постменопаузі з остеопорозом (плацебо [1,2 %, 50 з 4041] в порівнянні з препаратом Проліа® [1,5 %, 59 з 4050]); у чоловіків з остеопорозом (плацебо [0,8 %, 1 з 120] в порівнянні з препаратом Проліа® [0 %, 0 з 120]); у пацієнтів з раком грудної залози або простати, які отримують гормональну терапію (плацебо [1,7 %, 14 з 845] в порівнянні з препаратом Проліа® [1,4 %, 12 з 860]). Шкірні інфекції, що призвели до госпіталізації становили 0,1 % (3 з 4041) серед жінок в постменопаузі з остеопорозом, які отримували плацебо в порівнянні з 0,4 % (16 з 4050) серед жінок, які приймали препарат Проліа®. У більшості випадків це був целюліт. Інфекції шкіри, які повідомлялись як серйозні побічні реакції, були подібними у групі плацебо (0,6 %, 5 з 845) та групі, в якій приймали Проліа® (0,6 %, 5 з 860) в дослідженнях раку грудної залози і простати.
Остеонекроз щелепи
Випадки виникнення остеонекрозу щелепи повідомлялися рідко, у 16 хворих, в клінічних випробуваннях при остеопорозі та при раку грудної залози або раку простати у пацієнтів, які отримували гормональну терапію з загальною кількістю 23148 пацієнтів (див. розділ «Особливості застосування»). 13 з цих випадків сталися у жінок в постменопаузі з остеопорозом під час клінічного випробування фази III з продовженням подальшого лікування препаратом Проліа® до 10 років. Частота випадків виникнення остеонекрозу щелепи становила 0,04 % через 3 роки, 0,06 % через 5 років та 0,44 % через 10 років лікування препаратом Проліа®. Ризик виникнення остеопорозу щелепи зростав зі збільшенням періоду лікування препаратом Проліа®.
Атипові переломи стегна
У пацієнтів, яким застосовували препарат Проліа® згідно програми клінічних випробувань лікування остеопорозу, рідко повідомлялося про атипові переломи стегна (див. розділ «Особливості застосування»).
Дивертикуліт
В фазі III одного плацебо-контрольованого клінічного дослідження пацієнтів з раком простати, які отримували ADT спостерігали дисбаланс у побічних реакціях відносно дивертикуліту (1,2 % - у групі деносумабу, 0 % - у групі плацебо). Захворюваність дивертикулітом була порівнянна між групами лікування у жінок в постменопаузальному періоді або чоловіків з остеопорозом і у жінок з лікуванням неметастатичного раку молочної залози інгібітором ароматази.
Медикаментозна гіперчутливість
При постмаркетингових спостереженнях у пацієнтів, що отримували лікування препаратом Проліа®, спостерігалися рідкісні явища медикаментозної гіперчутливості, включаючи висипання, кропив'янку, набряк обличчя, еритему і анафілактичні реакції.
М'язово-скелетний біль
При постмаркетингових спостереженнях у хворих, які отримували препарат Проліа®, повідомляли щодо м'язово-скелетного болю, включаючи тяжкі випадки. У клінічних випробуваннях м'язово-скелетний біль спостерігали дуже часто в обох групах дослідження: лікування деносумабом та плацебо. Нечасто повідомляли щодо м'язово-скелетного болю, що призводив до припинення лікування у клінічному дослідженні.
Інші спеціальні групи пацієнтів
У клінічних дослідженнях пацієнти з тяжкою нирковою недостатністю (кліренс креатиніну < 30 мл/хв) або пацієнти, які отримують діаліз, піддаються більшому ризику гіпокальціємії за відсутності добавок кальцію. Адекватне споживання кальцію і вітаміну D має важливе значення у пацієнтів з тяжкою нирковою недостатністю або пацієнтів, які отримують діаліз (див. розділ «Особливості застосування»).
Повідомлення про підозрювані побічні реакції
Важливо повідомляти про підозрювані побічні реакції після реєстрації лікарського засобу. Це дає змогу продовжувати контролювати баланс користі/ризику лікарського засобу. Фахівців в галузі охорони здоров'я просять повідомляти про будь-які підозрювані побічні реакції через національну систему звітності.
Несумісність
Через відсутність досліджень сумісності, цей лікарський засіб не слід змішувати з іншими лікарськими засобами.
Термін придатності. 3 роки.
Умови зберігання.
Зберігати у холодильнику при температурі 2-8 °C в оригінальній упаковці. Після вилучення з холодильника зберігати при температурі не вище 25 °C в оригінальній упаковці. Не заморожувати. Не струшувати. Термін придатності після вилучення з холодильника - 30 днів. Зберігати в недоступному для дітей місці.
Упаковка
Попередньо заповнений шприц
Скляний попередньо заповнений шприц з голкою, закритою ковпачком, із захисним пристроєм від випадкового уколу голкою, що містить 1 мл розчину для ін'єкцій. По 1 попередньо заповненому шприцу з захисним пристроєм у блістері, поміщеному в картонну коробку,
або
скляний попередньо заповнений шприц з голкою, закритою ковпачком, що містить 1 мл розчину для ін'єкцій. По 1 попередньо заповненому шприцу в блістері або без блістера, поміщеному в картонну коробку.
Флакон
Скляний флакон із фторполімерною ламінованою еластомерною пробкою та алюмінієвою обкаткою з захисною пластиковою кришечкою, що містить 1 мл розчину для ін'єкцій. По 1 флакону в картонній коробці.
Категорія відпуску. За рецептом.
Виробник/заявник.
Амджен Європа Б.В., Нідерланди/Amgen Europe B.V., The Netherlands.
Місцезнаходження виробника та його адреса місця провадження діяльності
Мінервум 7061, 4817 ZK, Бреда, Нідерланди/
Minervum 7061, 4817 ZK, Breda, The Netherlands.
Інші медикаменти цього ж виробника
Форма: розчин для ін'єкцій, 60 мг/мл по 1 мл розчину в скляному попередньо наповненому шприці з голкою, закритою ковпачком, із захисним пристроєм від випадкового уколу голкою; по 1 попередньо заповненому шприцу з захисним пристроєм в блістері; по 1 блістеру в кортонній коробці; по 1 мл розчину в скляному попередньо заповненому шприці з голкою, закритою ковпачком; по 1 попередньо заповненому шприцу в блістері або без блістера, поміщеному в картонну коробку; по 1 мл розчину в скляному флаконі; по 1 флакону в картонній коробці
Форма: концентрат для розчину для інфузій, 20 мг/мл по 5 мл у флаконі; по 1 флакону в картонній коробці
Форма: розчин для ін'єкцій, 100 мкг/мл, по 0,3 мл у попередньо наповненому шприці з автоматичним запобіжником голки; по 1 шприцу у блістері; по 1 блістеру в коробці; по 0,3 мл у попередньо наповненому шприці; по 1 шприцу в коробці; по 0,3 мл у попередньо наповненому шприці; по 1 шприцу у блістері; по 1 блістеру в коробці
Форма: розчин для ін`єкцій, 140 мг/мл, по 1 мл у попередньо наповнених шприцах № 1, шприцах-ручках № 1 або № 2
Форма: розчин для ін'єкцій, 5 мг/мл; по 0,5 мл (2,5 мг) у флаконі, по 6 флаконів у картонній коробці; по 1 мл (5 мг) у флаконі, по 6 флаконів у картонній коробці; по 2 мл (10 мг) у флаконі, по 6 флаконів у картонній коробці