Іларіс
Реєстраційний номер: UA/14525/01/01
Імпортер: Новартіс Фарма АГ
Країна: ШвейцаріяАдреса імпортера: Ліхтштрассе 35, 4056 Базель, Швейцарія
Форма
порошок для розчину для ін`єкцій по 150 мг у флаконах № 1 або № 4
Склад
1 флакон містить 150 мг канакінумабу
Виробники препарату «Іларіс»
Країна: Швейцарія
Адреса: Шаффхаусерштрассе, 4332 Штейн, Швейцарія
Країна: Франція
Адреса: вул. де ля Шапель 26, 68330 Хюнінг, Франція
Країна: Швейцарія
Адреса: Ліхтштрассе 35, 4056 Базель, Швейцарія
Інструкція по застосуванню
для медичного застосування лікарського засобу
ІЛАРІС
(ILARIS®)
Склад
діюча речовина: canakinumab;
1 флакон містить 150 мг канакінумабу;
допоміжні речовини: сахароза, L-гістидин, L-гістидину гідрохлориду моногідрат, полісорбат 80.
Лікарська форма. Порошок для розчину для ін'єкцій.
Основні фізико-хімічні властивості: ліофілізований порошок білого кольору.
Фармакотерапевтична група. Інгібітори інтерлейкіну. Код АТC L04AC08.
Фармакологічні властивості.
Фармакодинаміка.
Механізм дії
Канакінумаб є моноклональним антитілом повністю людського походження ізотипу IgG1/κ до інтерлейкіну-1 бета (IL-1 бета). Канакінумаб специфічно зв'язується з високою спорідненістю з людським IL-1 бета і нейтралізує біологічну активність людського IL-1 бета, блокуючи його взаємодію з рецепторами IL-1, тим самим запобігаючи активації IL-1 бета-індукованого гена і продукування запальних медіаторів.
Фармакодинамічні ефекти
Періодичні синдроми, пов'язані з кріопірином
У клінічних дослідженнях у пацієнтів із періодичними синдромами, пов'язаними з кріопірином, у яких має місце неконтрольований викид IL-1 бета, спостерігається швидка реакція на терапію канакінумабом, тобто лабораторні показники, такі як високий рівень С-реактивного білка (СРБ) і сироваткового амілоїду А (САА), високий рівень нейтрофілів та тромбоцитів і лейкоцитоз, швидко нормалізувалися.
Системний ювенільний ідіопатичний артрит
Системний ювенільний ідіопатичний артрит є тяжким
аутозапальним вродженим захворюванням, спричиненим
прозапальними цитокінами, ключовим з яких є IL-1 бета.
Основні ознаки системного ювенільного ідіопатичного артриту
включають лихоманку, висип, гепатоспленомегалію,
лімфаденопатію, полісерозит і артрит.
Лікування канакінумабом спричиняло швидке і стійке зменшення як суглобових, так і системних проявів системного ювенільного ідіопатичного артриту зі значним скороченням кількості запалених суглобів, послабленням лихоманки і зниженням гостроти реактивного опору у більшості пацієнтів.
Подагричний артрит
Напад подагричного артриту спричинений кристалами
уратів (моногідрати мононатрієвих уратів) у суглобах і
навколишніх тканинах, які активізують резидентні макрофаги
для
утворення IL-1 бета через комплекс NALP3 інфламасоми.
Активація макрофагів і супутній підвищений викид IL-1 бета
призводить до гострої хворобливої запальної
реакції. Інші активатори природної імунної системи, такі як
ендогенні агоністи толл-подібних рецепторів, можуть робити
внесок у транскрипційну активацію гена IL-1 бета, ініціюючи
напад подагричного артриту. Після лікування канакінумабом
маркери запалення С-реактивний білок (СРБ) і сироватковий
амілоїд А (САА) з ознаками гострого запалення (наприклад
біль, набряк, почервоніння) в ураженому суглобі швидко
зникають.
Клінічна ефективність і безпека
Періодичні синдроми, пов'язані з кріопірином
Ефективність та безпека препарату Іларіс були продемонстровані у пацієнтів з різним ступенем тяжкості захворювання і різними фенотипами (у тому числі тяжких форм сімейного холодового аутозапального синдрому / сімейної холодової кропив'янки, синдрому Макла-Уельса і мультисистемного запального захворювання неонатального віку / хронічного дитячого неврологічного шкірно-артикулярного синдрому). Тільки пацієнти із підтвердженою мутацією NLRP3 були включені в ключове дослідження.
У фазі дослідження I/II препарат Іларіс мав швидкий початок дії: симптоми зникали або значно слабшали протягом одного дня. Лабораторні показники, такі як рівень СРБ і САА, рівень нейтрофілів і тромбоцитів, швидко нормалізувалися протягом декількох днів після введення Іларісу.
Ключове 48-тижневе багатоцентрове дослідження складалося з трьох частин: 8-тижневого відкритого періоду (частина I), 24-тижневого рандомізованого, подвійного сліпого плацебо-контрольованого періоду виведення (частина II) та 16-тижневого відкритого періоду (частина III). Метою дослідження було оцінити ефективність, безпеку і переносимість Іларісу (150 мг або 2 мг/кг кожні 8 тижнів) у пацієнтів з періодичними синдромами, пов'язаними з кріопірином.
- Частина I: повна клінічна відповідь і відповідь біомаркерів на Іларіс (визначається як глобальна оцінка лікаря щодо аутозапального захворювання і захворювання шкіри ≤ мінімуму та значення СРБ або САА < 10 мг/л) спостерігалися у 97 % пацієнтів і з'являлися через 7 днів від початку лікування. Значне поліпшення було відзначено у клінічній оцінці лікаря щодо активності аутозапалення: глобальна оцінка активності аутозапального захворювання, оцінка захворювання шкіри (кропив'янка, шкірний висип), біль у суглобах, міалгія, головний біль/ мігрень, кон'юнктивіт, втома/нездужання, оцінка інших пов'язаних симптомів, оцінка симптомів пацієнтом.
- Частина II: у період відміни основного дослідження первинна кінцева точка була визначена як частка пацієнтів з рецидивом захворювання/ повторним спалахом: 0 % пацієнтів із рецидивом при застосуванні Іларісу в порівнянні з 81 % пацієнтів, рандомізованих у групу плацебо.
- Частина III: пацієнти, які отримували плацебо в частині II, у яких відбувся рецидив і зберігалася клінічна і серологічна відповідь після вступу у відкриту частину подовження дослідження Іларісу.
Таблиця 1. Ефективність у фазі III ключового плацебо-контрольованого дослідження у період виведення (Частина II)
|
Іларіс N=3D15 n(%) |
Плацебо N=3D16 n(%) |
p-рівень |
|
Первинна кінцева точка (спалах)Кількість пацієнтів із рецидивом захворювання в частині II |
0 (0 %) |
13 (81 %) |
<0,001 |
Маркери запалення*С-реактивний білок, мг/л Сироватковий амілоїд А, мг/л |
1,10 (0,40) 2,27 (-0,20) |
19,93 (10,50) 71,09 (14,35) |
<0,001 0,002 |
|
*Середня зміна від початку частини II |
Проведено два відкритих, неконтрольованих, довготривалих дослідження фази III. Одне з них досліджувало безпеку, переносимість та ефективність канакінумабу у пацієнтів з періодичними синдромами, пов'язаними з кріопірином. Загальна тривалість лікування становить від 6 місяців до 2 років. Інше було відкритим дослідженням канакінумабу для оцінки ефективності і безпеки серед пацієнтів-японців із періодичними синдромами, пов'язаними з кріопірином, тривалістю 24 тижні, із фазою розширення до 48 тижнів. Основною метою було оцінити частку пацієнтів без рецидиву на 24-му тижні, у тому числі тих пацієнтів, у яких доза була збільшена.
За об'єднаним аналізом ефективності цих двох досліджень 65,6 % пацієнтів, які раніше не лікувалися канакінумабом, досягли повної відповіді при дозі 150 мг або 2 мг/кг, у той час як 85,2 % пацієнтів досягли повної ремісії при будь-якій дозі. Серед пацієнтів, які застосовували 600 мг або 8 мг/кг (або навіть вище), 43,8 % досягли повної відповіді. Кількість пацієнтів у віці від 2 до 4 років, які досягли повної відповіді (57,1 %), була менше порівняно із старшими дітьми та дорослими пацієнтами. Серед пацієнтів, які досягли повної відповіді, 89,3 % підтримували відповідь без рецидиву.
Досвід застосування окремим пацієнтам, які досягли повної відповіді після збільшення дози до 600 мг (8 мг/кг) кожні 8 тижнів, показує, що більш висока доза може бути корисною для пацієнтів, які не досягли повної відповіді або які не підтримують повну відповідь при рекомендованих дозах (150 мг або 2 мг/кг для пацієнтів ≥ 15 кг або ≤ 40 кг). Збільшена доза частіше вводилася пацієнтам віком від 2 до 4 років і пацієнтам із симптомами NOMID/CINCA у порівнянні з FCAS або MWS.
Педіатрична популяція
У дослідженнях періодичних синдромів, пов'язаних з кріопірином, брали участь загалом 80 педіатричних пацієнтів віком від 2 до 17 років (приблизно половині з них призначали дозу відповідно до маси тіла). В цілому, у педіатричних пацієнтів не було жодних клінічно значущих відмінностей в ефективності, безпеці та профілі переносимості Іларісу у порівнянні із загальною популяцією. Більшість педіатричних пацієнтів досягли поліпшення клінічних симптомів та об'єктивних маркерів запалення (наприклад САА і СРБ).
Ефективність, безпеку та переносимість препарату
Іларіс оцінювали у відкритому 56-тижневому дослідженні за
участю пацієнтів дитячого (≤4 років) віку з періодичними
синдромами, пов'язаними з кріопірином. Оцінювали сімнадцять
пацієнтів (включаючи 6 пацієнтів віком до
2 років), застосовуючи початкові дози, розраховані на
основі маси тіла - 2-8 мг/кг.
В дослідженні також оцінювали вплив
канакінумабу на утворення антитіл до стандартних дитячих
вакцин. Ніяких відмінностей в безпеці або ефективності не
спостерігалося у пацієнтів віком
до 2 років у порівнянні з пацієнтами
віком від 2 років. Всі пацієнти, які
отримували неживі стандартні дитячі вакцини
(N =3D 7),
мали захисні рівні антитіл.
Системний ювенільний ідіопатичний артрит
Ефективність препарату Іларіс для лікування активного
системного ідіопатичного ювенільного артриту оцінювали у
двох базових дослідженнях (G2305 і G2301). Включені до
дослідження пацієнти були віком від 2 до 20 років (середній
вік 8,5 року і середня тривалість хвороби
3,5 року на початку дослідження) і мали
активне захворювання, що визначалося за наявністю
2 і більше суглобів з активним
артритом, лихоманкою і підвищеним СРБ.
Дослідження G2305
Дослідження G2305 було рандомізоване, подвійне сліпе,
плацебо-контрольоване, 4-тижневе.
У ньому вивчали короткострокову ефективність препарату
Іларіс у 84 пацієнтів, рандомізованих для отримання однієї
дози 4 мг/кг (до 300 мг) препарату Іларіс або плацебо.
Основною метою було встановлення частки пацієнтів, яка на
15-й день досягла мінімального поліпшення на 30 %
відповідно до критеріїв педіатричної Американської колегії
ревматологів (АКР). Ці критерії біли адаптовані для
можливості включення пацієнтів без лихоманки. Лікування
препаратом Іларіс поліпшило усі показники АКР в порівнянні
з плацебо на 15-й та 29-й день (таблиця 2).
Таблиця 2. Педіатричні показники АКР і статус захворювання на 15-й та 29-й день
|
15-й день |
29-й день |
|||
|
Іларіс N=3D43 |
Плацебо N=3D41 |
Іларіс N=3D43 |
Плацебо N=3D41 |
|
|
AКР30 |
84 % |
10 % |
81 % |
10 % |
|
AКР50 |
67 % |
5 % |
79 % |
5 % |
|
AКР70 |
61 % |
2 % |
67 % |
2 % |
|
AКР90 |
42 % |
0 % |
47 % |
2 % |
|
AКР100 |
33 % |
0 % |
33 % |
2 % |
|
Неактивне захворювання |
33 % |
0 % |
30 % |
0 % |
|
Різниця в лікуванні для всіх показників АКР була значною (p ≤ 0,0001) |
Результати адаптованих педіатричних показників АКР, які включали системні та артритичні компоненти, узгоджувалися із загальними результатами показників АКР. На 15-й день середня зміна від вихідного рівня у кількості суглобів з активним артритом і обмежений діапазон руху становили −67 % і −73 % для Іларісу (N =3D 43) відповідно, в порівнянні з медіаною зміни 0 % і 0 % у групі плацебо (N =3D 41). Середня зміна у шкалі болю у пацієнта (0-100 мм візуальної аналогової шкали) на 15-й день була 50,0 мм для Іларісу (N =3D 43), у порівнянні з +4,5 мм для плацебо (N =3D 25). Середня зміна показника болю серед пацієнтів була узгодженою на 29-й день.
Дослідження G2301
Дослідження G2301 було рандомізоване, подвійне сліпе, плацебо-контрольоване дослідження з вивчення профілактики загострень при терапії препаратом Іларіс. Дослідження складалося з двох частин з двома незалежними первинними кінцевими точками (успішне зниження стероїдів і часу до запалення). У першій частині (відкрита) 177 пацієнтів було включено і отримувало 4 мг/кг (до 300 мг) препарату Іларіс, що вводили кожні 4 тижні протягом до 32 тижнів. Пацієнти у частині II (подвійна сліпа) отримували Іларіс 4 мг/кг або плацебо кожні 4 тижні до спостереження 37 спалахів.
Зменшення дози кортикостероїдів
Зі 128 пацієнтів, які брали приймали кортикостероїди та брали участь в першій частині дослідження, 92 пацієнти намагались знизити дозу кортикостероїдів. П'ятдесят сім (62%) з них змогли значно знизити дозу кортикостероїдів, а 42 (46%) - припинили застосування кортикостероїдів.
Час до першого спалаху
У пацієнтів, які приймали препарат Іларіс в другій частині дослідження, спостерігалось зниження ризику спалаху захворювання на 64% порівняно з групою плацебо (співвідношення ризиків 0,36; 95% ДІ: 0,17 - 0,75; p=3D0,0032). У шістдесяти трьох зі 100 пацієнтів, які брали участь в другій частині дослідження в групі плацебо або канакінумабу, не спостерігалось спалаху захворювання протягом періоду спостереження (максимально до 80 тижнів).
Результати досліджень G2305 та G2301, пов'язані зі здоров'ям і якістю життя
На фоні лікування препаратом Іларіс спостерігалось клінічно значуще покращення фізичної функції і якості життя пацієнтів. Під час дослідження G2305 за даними анкети оцінки стану здоров'я дітей за методом найменших квадратів покращення становило 0,69 в групі препарату Іларіс в порівнянні з групою плацебо, що в 3,6 раза перевищує мінімальну клінічно значущу відмінність 0,19 (р =3D 0,0002). Середнє покращення в порівнянні з вихідним рівнем наприкінці другої частини дослідження G2301 становило 0,88 (79%). Статистично значуще покращення за показниками анкети оцінки стану здоров'я дітей PF50 спостерігалося в групі препарату Іларіс в порівнянні з групою плацебо у дослідженні G2305 (фізичний стан р =3D 0,0012; психосоціальне благополуччя р =3D 0,0017).
Об'єднаний аналіз ефективності
Дані перших 12 тижнів лікування препаратом Іларіс, отримані під час досліджень G2305, G2301, а також розширеного дослідження, були об'єднані, для оцінки ефективності. Ці дані свідчать про однакові покращення в порівнянні з вихідним рівнем адаптованих педіатричних показників АКР та їх компонентів вже до 12-го тижня порівняно з тими, що спостерігалися в плацебо-контрольованому дослідженні (G2305). На 12-й тиждень адаптовані педіатричні показники АКР30, 50, 70, 90 і 100 становили: 70%, 69%, 61%, 49% і 30% відповідно, тоді як 28% пацієнтів мали неактивне захворювання (N =3D 178).
Ефективність, що спостерігалась під час проведення досліджень G2305 і G2301, підтримувалась в довгостроковому відкритому розширеному дослідженні, яке наразі продовжується (існуючі дані за період спостереження з медіаною 49 тижнів). У цьому дослідженні 25 пацієнтів, які мали сильну реакцію за даними АКР протягом щонайменше 5 місяців, знижували дозу препарату Іларіс до 2 мг/кг кожні 4 тижні і підтримували педіатричну ACR100 відповідь під час застосування зниженої дози (медіана 32 тижні, 8-124 тижні).
Хоча результати є обмеженими, дані клінічних досліджень свідчать про те, що пацієнти, які не реагують на лікування тоцилізумабом або анакінрою, можуть реагувати на лікування канакінумабом.
Подагричний артрит
Ефективність Іларісу для лікування гострих нападів подагричного артриту була продемонстрована у двох багатоцентрових, рандомізованих, подвійних сліпих, активних контрольованих дослідженнях у пацієнтів з частим проявом подагричного артриту (3 або більше нападів протягом попередніх 12 місяців) при неможливості застосування НПЗП або колхіцину (у зв'язку з протипоказаннями, непереносимістю або недостатньою ефективністю). Дослідження тривали 12 тижнів з наступним 12-тижневим подвійним сліпим розширенням. Загалом 225 пацієнтам застосовували Іларіс підшкірно у дозі 150 мг і 229 пацієнтів отримували внутрішньом'язово триамцинолону ацетонід (ТА) у дозі 40 мг на момент початку дослідження і після повторення нападу. Середня кількість нападів подагричного артриту протягом попередніх 12 місяців становила 6,5. Більше 85 % пацієнтів мали супутні захворювання, в тому числі артеріальну гіпертензію (60 %), цукровий діабет (15 %), ішемічну хворобу серця (12 %) і хронічні захворювання нирок стадії ≥ 3 (25 %). Приблизно одна третина пацієнтів, включених у дослідження (76 [33,8 %] в групу Іларісу і 84 [36,7 %] в групу триамцинолону), не могли застосовувати НПЗП і колхіцин (непереносимість, протипоказання або відсутність реакції). Супутня терапія для зниження уратів (ULT) застосовувалася 42 % пацієнтів при включенні у дослідження.
Складовими первинними кінцевими точками були: (I) інтенсивність болю при подагричному артриті (за візуальною аналоговою шкалою, VAS ) через 72 години після введення дози, і (II) час першого нового нападу подагричного артриту.
У загальній популяції дослідження інтенсивність болю була статистично значно нижче для Іларісу 150 мг порівняно з триамцинолону ацетонідом через 72 години. Іларіс також знижує ризик подальших нападів (див. таблицю 3).
Результати ефективності у підгрупі пацієнтів, які не можуть застосовувати НПЗП і колхіцин, і тих, хто приймали ULT, не отримали результату від ULT або мали протипоказання до ULT (N =3D 101), узгоджувалися із дослідженням загальної популяції із статистично значущими відмінностями в порівнянні з триамцинолону ацетонідом щодо інтенсивності болю через 72 години (−10,2 мм, р =3D 0,0208 ) і зниженні ризику подальших нападів (співвідношення ризиків 0,39, р =3D 0,0047 на 24-му тижні).
Результати ефективності для скороченої підгрупи, обмеженої пацієнтами, які застосовували ULT (N =3D 62), представлені в таблиці 4. Лікування Іларісом спричиняло зменшення болю і зниження ризику подальших нападів у пацієнтів, яким застосовують ULT і які не можуть застосовувати НПЗП і колхіцин, хоча різниця у лікуванні порівняно з триамцинолону ацетонідом була менш вираженою, ніж у загальній популяції дослідження.
Таблиця 3. Ефективність у загальній популяції дослідження та підгрупі пацієнтів, яким застосовують ULT і які не можуть застосовувати НПЗП або колхіцин
|
Кінцева точка ефективності |
Загальна популяція дослідження N=3D454 |
Пацієнти, які не можуть застосовувати НПЗП та колхіцин, яким застосовують ULT N=3D62 |
|
Лікування нападів подагричного артриту (інтенсивність болю (VAS) через 72 години) |
||
|
Оцінка середньої різниці методом найменших квадратів для триамцинолону ацетоніду СІ p-рівень, 1-сторонній |
−10,7 (−15,4, −6,0) p<0,0001* |
−3,8 (−16,7, 9,1) p=3D0,2798 |
|
Ризик зниження наступних нападів подагричного артриту, що оцінюється за часом до першого спалаху (24 тижні) |
||
|
Співвідношення ризику для триамцинолону ацетоніду СI p-рівень, 1-сторонній |
0,44 (0,32, 0,60) p<0,0001* |
0,71 (0,29, 1,77) р=3D0,2337 |
|
*Позначає значущий p-рівень ≤0,025. |
Результати дослідження безпеки показали збільшення кількості випадків несприятливих подій після застосування канакінумабу в порівнянні з триамцинолону ацетонідом: 66 % проти 53 % пацієнтів, у яких виникали будь-які негативні події, і 20 % проти 10 % пацієнтів, у яких виникали випадки інфекцій, протягом 24 тижнів.
Пацієнти літнього віку
В цілому, ефективність, безпека та профіль переносимості Іларісу у літніх пацієнтів (≥ 65 років) були порівнянними з такими у пацієнтів віком до 65 років.
Пацієнти на терапії для зниження уратів (ULT)
У клінічних дослідженнях Іларіс безпечно застосовували разом із ULT. У загальній популяції дослідження пацієнти на ULT мали менш виражене зменшення болю і зниження ризику подальших нападів подагричного артриту порівняно з пацієнтами, які не проходили ULT.
Імуногенність
Жодних анафілактичних реакцій не спостерігалося у пацієнтів, які отримували Іларіс.
Антитіла проти препарату Іларіс спостерігалися приблизно у 1,5 %, і 2 % пацієнтів, які отримували Іларіс для лікування періодичних синдромів, пов'язаних з кріопірином, і подагричного артриту відповідно.
Цей лікарський засіб було дозволено для пацієнтів з періодичними синдромами, пов'язаними з кріопірином, при "виняткових обставинах". Це означає, що через рідкість захворювання не вдалося отримати повну інформацію щодо цього лікарського засобу. Європейське агентство з лікарських засобів розгляне будь-яку нову інформацію, і коротка характеристика препарату буде оновлюватися в міру необхідності.
Педіатрична популяція
Європейське агентство з лікарських засобів відклало зобов'язання представляти результати досліджень Іларісу в одному або декількох піддослідженнях педіатричної популяції з періодичними синдромами, пов'язаними з кріопірином. Європейське агентство з лікарських засобів відклало зобов'язання представляти результати досліджень Іларісу в усіх піддослідженнях педіатричної популяції при подагричному артриті.
Фармакокінетика.
Періодичні синдроми, пов'язані з кріопірином (CAPS)
Всмоктування
Пікова сироваткова концентрація канакінумабу
(Cmax) спостерігалася
приблизно через 7 днів після одноразового підшкірного
введення 150 мг дорослим пацієнтам з
CAPS. Середній період напіввиведення
становив 26 днів. Середні значення
Cmax та
AUCinf після введення
одноразової підшкірної дози 150 мг типовому дорослому
пацієнту з CAPS (70 кг) становили
15,9 мкг/мл і 708 мкг* д/мл. Абсолютна біодоступність при
підшкірному введенні канакінумабу за оцінками становить 66
%. Параметри експозиції (наприклад AUC
і Cmax)
збільшувалися пропорційно дозі в діапазоні доз від 0,30 до
10,0 мг/кг, введених у вигляді внутрішньовенної інфузії,
або від 150 до 600 мг у вигляді підшкірної ін'єкції.
Прогнозовані значення стаціонарної експозиції
(Cmin,ss, Cmax,ss,
AUC,ss,8w) після підшкірного
введення 150 мг (або 2 мг/кг, відповідно)
кожні 8 тижнів були дещо вищіу ваговій
категорії 40-70 кг (6,6 µg/мл, 24,3 µg/мл, 767
µg*d/мл) порівняно з ваговими
категоріями < 40 кг (4,0 µg/мл, 19,9 µg/мл,
566 µg*d/мл) і
> 70 кг (4,6 µg/мл, 17,8 µg/мл, 545 µg*d/мл).
Очікуваний коефіцієнт накопичення становив
1,3 раза після 6 місяців підшкірного введення 150 мг
канакінумабу кожні 8 тижнів.
Розподіл
Канакінумаб зв'язується з IL-1 бета в сироватці крові. Об'єм розподілу (Vss) канакінумабу варіює залежно від маси тіла. За розрахунками, він становить 6,2 літра у пацієнтів із періодичними синдромами, пов'язаними з кріопірином, маса тіла яких 70 кг.
Виведення
Видимий кліренс (CL/F) канакінумабу збільшується з масою тіла. Він за оцінками становить 0,17 л/день у пацієнтів з періодичними синдромами, пов'язаними з кріопірином з масою тіла 70 кг та 0,11 л/день у пацієнтів з системним ювенільним ідіопатичним артритом маса тіла яких 33 кг.
Не було жодних ознак прискореного кліренсу або залежних від часу змін фармакокінетичних властивостей канакінумабу після повторного введення. Після корекції маси тіла жодних статевих або вікових фармакокінетичних відмінностей не спостерігалося.
Системний ювенільний ідіопатичний артрит
Біодоступність у пацієнтів з системним ювенільним ідіопатичним артритом окремо не визначалась. Видимий кліренс на кілограм маси тіла (CL/F на кг) порівнювали у групах пацієнтів із системним ювенільним ідіопатичним артритом та періодичними синдромами, пов'язаними з кріопірином (0,004 л/добу/кг). Видимий об'єм розподілу на кілограм маси тіла (V/F на кг) становив 0,14 л/кг.
Після введення повторних доз 4 мг/кг кожні 4 тижні коефіцієнт кумуляції канакінумабу був у 1,6 раза вищим у пацієнтів із системним ювенільним ідіопатичним артритом. Стабільний стан досягався через 110 днів. Загальні прогнозовані середні значення (±СВ) Cmin,ss, Cmax,ss та AUC,ss4w становили 14,7±8,8 мкг/мл, 36,5 ± 14,9 мкг/мл та 696,1 ± 326.5 мкг*добу/мл відповідно.
AUCss4w в кожній віковій групі становив 692, 615, 707 та 742 мкг*добу/мл у пацієнтів віком 2‑3, 4‑5, 6‑11 та 12‑19 років відповідно. При стратифікації за масою тіла відмічалась нижня (30-40%) медіана експозиції Cmin,ss (11,4 порівняно з 19 мкг/мл) та AUCss (594 порівняно з 880 мкг*добу/мл) у категорії пацієнтів з меншою масою тіла (≤ 40 кг) порівняно з пацієнтами з високою масою тіла (> 40 кг).
Пацієнти із подагричним артритом
Біодоступність у пацієнтів із подагричним артритом не була визначена. Видимий кліренс на кілограм маси тіла (CL/F на кг) порівнювали між групами пацієнтів з подагричним артритом і CAPS (0,004 л/добу/кг). Середня експозиція у типового пацієнта із подагричним артритом (93 кг) після одноразової підшкірної дози 150 мг (Cmax: 10,8 мкг/мл і AUCinf: 495 мкг* д/мл) була нижчою, ніж у типових пацієнтів з CAPS при вазі 70 кг (15,9 мкг/мл і 708 мкг* д/мл). Це узгоджується із спостережуваним збільшенням CL/F з масою тіла.
Очікуваний коефіцієнт кумуляції був у 1,1 раза вищим після підшкірного введення канакінумабу у дозі 150 мг кожні 12 тижнів.
Діти
Пік концентрації канакінумабу був досягнений через 2−7 днів після одноразового підшкірного введення канакінумабу 150 мг або 2 мг/кг у педіатричних хворих 4-х річного віку і старших. Період напіврозпаду коливався в діапазоні від 22,9 до 25,7 дня, подібно до такого у дорослих. На основі аналізу фармакокінетичного моделювання фармакокінетика канакінумабу у дітей у віці від 2 до 4 років була аналогічною такій у хворих віком від 4 років.
Було визначено, що при підшкірному введенні ступінь абсорбції знижувався з віком та прискорюється у молодих пацієнтів. Відповідно, Tmax був коротшим (3,6 дня) у молодих пацієнтів з системним ювенільним ідіопатичним артритом (2‑3 роки) порівняно з більш старшими пацієнтами з системним ювенільним ідіопатичним артритом (12‑19 років; Tmax: 6 днів). Жодного негативного впливу на біодоступність (AUCss) не виявлено.
Додатковий фармакокінетичний аналіз показав, що фармакокінетика канакінумабу у 6 пацієнтів віком до 2 років з періодичними синдромами, пов'язаними з кріопірином, була подібною до фармакокінетики у дітей віком 2-4 роки. Популяційне моделювання фармакокінетики свідчить, що прогнозовані рівні експозиції після прийому дози 2 мг/кг були порівняними у пацієнтів дитячого віку з періодичними синдромами, пов'язаними з кріопірином, однак на 40% меншими у пацієнтів з дуже низькою масою тіла, наприклад 10 кг, порівняно з дорослими пацієнтами
(доза 150 мг/кг). Це узгоджується з більш високими рівнями експозиції у групах пацієнтів з періодичними синдромами, пов'язаними з кріопірином, що мають вищу масу тіла.
Фармакокінетика є однаковою у дітей з періодичними синдромами, пов'язаними з кріопірином, та системним ювенільним ідіопатичним артритом.
Пацієнти літнього віку
Жодних змін фармакокінетичних параметрів на основі кліренсу або об'єму розподілу не було виявлено у літніх пацієнтів і дорослих пацієнтів віком до 65 років.
Доклінічні дані з безпеки
Доклінічні дані не показали специфічної небезпеки для людей на основі даних досліджень перехресної реактивності, застосування повторних доз, імунотоксичності, дослідження репродуктивної та ювенільної токсичності, виконаних із канакінумабом або мишачими антимишачими антитілами IL-1 бета.
Оскільки канакінумаб зв'язується із мавп'ячим (С. jacchus) і людським IL-1 бета з подібною спорідненістю, безпека канакінумабу вивчалася на мавпах. Жодних побічних ефектів канакінумабу не було відзначено після введення мавпам препарату два рази на тиждень протягом 26 тижнів або при дослідженні токсичності ембріофетального розвитку у вагітних мавп. Концентрації в плазмі, які добре переносяться у тварин, перевищують щонайменше у 42 рази (Cmax) і 78 разів (CAVG) концентрації в плазмі у педіатричних пацієнтів з CAPS (маса тіла 10 кг), яким застосовували клінічні дози канакінумабу до 8 мг/кг підшкірно кожні 8 тижнів. Крім того, антитіла до канакінумабу не були виявлені у цих дослідженнях. Жодної неспецифічної тканинної перехресної реактивності не було продемонстровано після нанесення канакінумабу на нормальні тканини людини.
Формальні дослідження канцерогенності канакінумабу не проводилися.
У дослідженні ембріофетального розвитку у мавп канакінумаб не показав материнської токсичності, ембріотоксичної або тератогенної дії при введенні протягом органогенезу.
Жодних побічних ефектів з мишачими антимишачими антитілами IL-1 бета не було відзначено у цілому ряді репродуктивних досліджень та досліджень на ювенільних мишах. Антимишачі IL-1 бета не чинили побічних ефектів на плід або на ріст новонародженого при введенні матері на пізніх термінах вагітності, під час пологів і годування груддю. Високі дози, що застосовували у цих дослідженнях, були максимально ефективними з точки зору пригнічення і активності IL-1 бета.
Імунотоксикологічні дослідження на мишах з мишачими антимишачими антитілами IL-1 бета показали, що нейтралізація IL-1 бета не має жодного впливу на імунологічні показники і не спричиняє порушення імунної функції у мишей.
Клінічні характеристики
Показання
Періодичні синдроми, пов'язані з кріопірином
Лікування періодичних синдромів, пов'язаних з кріопірином, у дорослих, підлітків і дітей віком від 2 років з масою тіла 7,5 кг або вище, в тому числі:
• синдрому Макла−Уельса;
• мультисистемного запального захворювання неонатального віку/ хронічного дитячого неврологічного шкірно-артикулярного синдрому;
• тяжких форм сімейного холодового аутозапального синдрому/ сімейної холодової кропив'янки із симптомами, що не характерні для кропив'янки, пов'язаної з холодом.
Системний ювенільний ідіопатичний артрит
Лікування активного системного ювенільного ідіопатичного артриту у пацієнтів віком від 2 років, у яких спостерігалася неадекватна відповідь на попередню терапію нестероїдними протизапальними препаратами (НПЗП) і системними кортикостероїдами. Іларіс можна застосовувати як монотерапію або в комбінації з метотрексатом.
Подагричний артрит
Симптоматичне лікування дорослих пацієнтів з частими нападами подагричного артриту (не менше 3 нападів протягом попередніх 12 місяців) у випадках, коли нестероїдні протизапальні препарати (НПЗП) і колхіцин протипоказані, не переносяться або не забезпечують адекватного ефекту, і коли призначення повторних курсів лікування кортикостероїдами не є прийнятним.
Протипоказання
Підвищена чутливість до діючої речовини або до будь-якої з допоміжних речовин. Активні, тяжкі інфекції.
Взаємодія з іншими лікарськими засобами та інші види взаємодій
Взаємодії Іларісу з іншими лікарськими засобами не були оцінені в офіційних дослідженнях.
Збільшення кількості випадків серйозних інфекцій було пов'язано із введенням іншого блокатора IL-1 в комбінації з інгібіторами фактора некрозу пухлин (ФНП). Застосування Іларісу з інгібіторами ФНП не рекомендується, оскільки це збільшує ризик серйозних інфекцій.
Активність печінкових ферментів CYP450 може бути пригнічена цитокінами, які стимулюють хронічне запалення, такими як інтерлейкін-1 бета (IL-1 бета). Таким чином, активність CYP450 може бути змінена при проведенні потужної інгібуючої терапії цитокінами, наприклад при введенні канакінумабу. Це має клінічне значення для субстратів CYP450 з вузьким терапевтичним індексом, коли доза корегується індивідуально. На початку застосування канакінумабу з цим типом лікарського засобу потрібно провести терапевтичний моніторинг ефекту або концентрації активної речовини і при необхідності відкоригувати дозу.
Відсутні дані щодо дії живих вакцин або вторинної передачі інфекції з живими вакцинами у пацієнтів, які застосовують Іларіс. Таким чином, живі вакцини не слід вводити одночасно з Іларісом, за винятком випадків, коли переваги явно перевищують ризики. Якщо вакцинацію живими вакцинами призначають після початку лікування Іларісом, рекомендується зробити перерву не менше 3 місяців після останньої ін'єкції Іларісу і перед наступною ін'єкцією.
Результати дослідження, проведеного серед здорових дорослих добровольців, показали, що одна доза 300 мг препарату Іларіс не впливає на індукцію і збереження відповіді антитіл після вакцинації проти грипу або менінгококовою вакциною на основі глікозильованого білка.
Результати 56-тижневого відкритого дослідження пацієнтів з періодичними синдромами, пов'язаними з кріопірином, віком до 4 років показали, що у всіх пацієнти, які отримали неживі вакцини, які є стандартом дитячої вакцинації, були вироблені захисні рівні антитіл.
Особливості застосування
Інфекції
Застосування Іларісу було пов'язане зі збільшенням кількості випадків серйозних інфекцій. Тому пацієнти повинні перебувати під ретельним наглядом щодо симптомів інфекцій під час і після лікування препаратом Іларіс. Лікарі повинні проявляти обережність при застосуванні препарату Іларіс пацієнтам з інфекціями, повторними інфекціями в анамнезі або наявністю станів, які можуть призвести до інфекцій.
Лікування періодичних синдромів, пов'язаних з кріопірином, та системного ювенільного ідіопатичного артриту
Іларіс не слід застосовувати під час активної інфекції, що вимагає медичного втручання.
Лікування подагричного артриту
Іларіс не слід застосовувати під час активної інфекції.
Одночасне застосування препарату Іларіс з інгібіторами фактору некрозу пухлини (ФНП) не рекомендується, оскільки це збільшує ризик серйозних інфекцій.
Поодинокі випадки незвичайних або опортуністичних інфекцій (включаючи аспергільоз, атипові мікобактеріальні інфекції, оперізуючий лишай) було зареєстровано під час лікування препаратом Іларіс. Однак причинний зв'язок препарату Іларіс з цими подіями не може бути виключений.
Близько 12 % пацієнтів із періодичними синдромами, пов'язаними з кріопірином, при проведенні туберкулінової шкірної проби (PDD) у клінічних дослідженнях мали позитивний результат, у той час коли їм застосовували Іларіс без клінічних ознак прихованої або активної туберкульозної інфекції.
Невідомо, чи підвищує застосування інгібіторів інтерлейкіну-1 (IL-1), таких як Іларіс, ризик реактивації туберкульозу. До початку терапії всіх пацієнтів слід перевірити на наявність активного і латентного туберкульозу. Лікарю необхідно детально ознайомитися з історією хвороби. У всіх пацієнтів потрібно провести відповідні скринінгові тести (наприклад туберкулінова шкірна проба, аналіз на вивільнення інтерферону гамма або рентген грудної
клітки). Слід ретельно спостерігати за пацієнтами стосовно симптомів туберкульозу під час і після лікування препаратом Іларіс. Пацієнт повинен знати, що якщо симптоми, що вказують на туберкульоз (наприклад постійний кашель, втрата ваги, субфебрильна температура), з'являються під час терапії препаратом Іларіс, йому необхідно звернутися до лікаря. Якщо проба Манту позитивна, особливо у пацієнтів з високим ризиком, слід розглянути альтернативні способи скринінгу туберкульозної інфекції.
Нейтропенія та лейкопенія
Нейтропенія (абсолютна кількість нейтрофілів [АКН] < 1,5 × 109/л) та лейкопенія спостерігалися при застосуванні лікарських засобів, які інгібують IL-1, в тому числі Іларісу. Лікування препаратом Іларіс не слід розпочинати у пацієнтів з нейтропенією або лейкопенією. Рекомендується оцінювати рівень клітин білої крові, в тому числі число нейтрофілів, до початку лікування і через 1 та 2 місяці після початку. Для терапії хронічних пацієнтів або повторної терапії пацієнтів також рекомендується періодично оцінювати рівень білих клітин крові під час лікування. Якщо пацієнт входить у нейтропенічний або лейкопенічний стан, слід пильно стежити за рівнем білих клітин крові та розглянути необхідність припинення лікування.
Злоякісні новоутворення
Злоякісності новоутворення були зареєстровані у пацієнтів, які застосовували Іларіс. Ризик розвитку злоякісних пухлин при застосуванні антиінтерлейкінів IL-1 невідомий.
Реакції гіперчутливості
Були зареєстровані випадки, що вказують на реакції гіперчутливості при застосуванні Іларісу. Більшість із цих випадків мали легкий ступінь тяжкості. Під час клінічної розробки Іларісу у більш ніж 2300 пацієнтів анафілактоїдні або анафілактичні реакції не відзначалися. Однак ризик тяжких реакцій гіперчутливості, що не рідкість для білкових ін'єкцій, не може бути виключений.
Функція печінки
Короткотривалі та безсимптомні випадки підвищення рівня сироваткових трансаміназ або білірубіну були зареєстровані у клінічних випробуваннях.
Вакцинація
Дані щодо ризику вторинної передачі інфекції з живими (ослабленими) вакцинами у пацієнтів, які застосовують Іларіс, відсутні. Таким чином, живі вакцини не слід вводити одночасно з препаратом Іларіс, крім випадків, коли переваги значно переважують ризики.
До початку терапії Іларісом дорослим пацієнтам і дітям рекомендовано отримати усі щеплення, при необхідності, в тому числі пневмококову вакцину та інактивовану грипозну вакцину.
Мутація в гені NLRP3 у пацієнтів з періодичними синдромами, пов'язаними з кріопірином
Клінічний досвід щодо пацієнтів з періодичними синдромами, пов'язаними з кріопірином, без підтвердженої мутації в гені NLRP3 обмежений.
Синдром активації макрофагів у пацієнтів з системним ювенільним ідіопатичним артритом
Синдром активації макрофагів - це відомий стан, що загрожує життю, який може розвиватися у пацієнтів з ревматичними захворюваннями, зокрема у пацієнтів із системним ювенільним ідіопатичним артритом. У разі розвитку синдрому активації макрофагів або підозри на нього оцінку та лікування слід розпочати якомога скоріше. Лікарям слід уважно ставитися до симптомів інфекції або погіршення перебігу системного ювенільного ідіопатичного артриту, відомих як пусковий механізм для синдрому активації макрофагів. Дані клінічних досліджень вказують на те, що препарат Іларіс, ймовірно, не збільшує ризик розвитку синдрому активації макрофагів у пацієнтів з системним ювенільним ідіопатичним артритом, однак не дозволять зробити остаточних висновків.
Застосування у період вагітності або годування груддю.
Вагітність
Дані щодо застосування препарату Іларіс вагітним жінкам обмежені. Досліди на тваринах не вказують на прямий або опосередкований несприятливий вплив препарату на репродуктивну функцію. Ризик для плода/матері невідомий. Жінки повинні використовувати ефективні засоби контрацепції під час лікування Іларісом і протягом 3 місяців після введення останньої дози. Препарат Іларіс слід застосовувати під час вагітності тільки у випадках, якщо очікувана користь для матері виправдовує потенційний ризик для плода.
Годування груддю
Невідомо, чи канакінумаб екскретується у грудне молоко людини. Питання про застосування препарату Іларіс жінкам, які годують груддю, слід розглядати тільки у тому випадку, якщо очікувана користь для жінки більша, ніж будь-який ризик для дитини.
Фертильність
Дослідження потенційного впливу Іларісу на фертильність у людей не були проведені.
Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами.
Іларіс може впливати на здатність керувати автотранспортом або працювати з механізмами, тому під час застосування препарату рекомендовано утриматися від керування транспортними засобами та іншої діяльності, що потребує концентрації уваги.
Спосіб застосування та дози
Періодичні синдроми, пов'язані з кріопірином, та системний ювенільний ідіопатичний артрит
Лікування слід розпочинати за призначенням та під
наглядом лікаря, який має досвід
у діагностиці та лікуванні відповідних станів.
Після належної підготовки щодо техніки ін'єкційного введення пацієнти або іхні опікуни можуть самостійно вводити Іларіс, якщо лікар визначить, що це доцільно і необхідно з медичної точки зору.
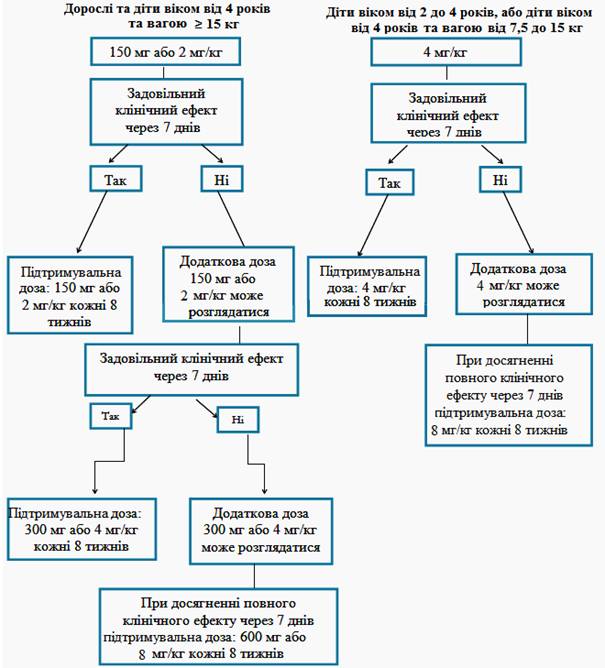
Рекомендовані початкові дози препарату Іларіс для пацієнтів з періодичними синдромами, пов'язаними з кріопірином, дорослих та дітей віком від 2 років.
Дорослі та діти віком від 4 років:
· 150 мг для пацієнтів із вагою тіла > 40 кг;
· 2 мг/кг для пацієнтів із вагою тіла ≥ 15 кг та ≤ 40 кг;
· 4 мг/кг для пацієнтів із вагою тіла ≥7,5 кг та <15 кг.
Діти від 2 до 4 років:
· 4 мг/кг для пацієнтів із вагою тіла ≥ 7,5 кг.
Ці дози вводять кожні вісім тижнів у вигляді разової дози шляхом підшкірної ін'єкції.
Якщо задовільного клінічного ефекту (зникнення висипу та інших загальних симптомів) при початковій дозі 150 мг або 2 мг/кг не було досягнуто через 7 днів після початку лікування, можливе введення другої дози препарату Іларіс 150 мг або 2 мг/кг. При досягненні клінічного ефекту від лікування необхідно підтримувати посилений режим дозування 300 мг або 4 мг/кг кожні 8 тижнів. Якщо задовільний клінічний ефект не був досягнутий через 7 днів після цього збільшення дози, можливе введення третьої дози препарату Іларіс 300 мг або 4 мг/кг. При досягненні повного клінічного ефекту від лікування необхідно підтримувати посилений режим дозування 600 мг або 8 мг/кг кожні 8 тижнів на основі індивідуальної клінічної оцінки.
Якщо задовільний клінічний ефект при початковій дозі 4 мг/кг не був досягнутий через 7 днів після початку лікування, можливе введення другої дози препарату Іларіс 4 мг/кг. При досягненні повного клінічного ефекту від лікування необхідно підтримувати посилений режим дозування 8 мг/кг кожні 8 тижнів на основі індивідуальної клінічної оцінки.
Клінічний досвід застосування доз з інтервалом менш ніж 4 тижні та застосування доз вище 600 мг або 8 мг/кг обмежений.
Системний ювенільний ідіопатичний артрит
Рекомендована доза препарату Іларіс для пацієнтів із системним ювенільним ідіопатичним артритом із масою тіла ≥ 7,5 кг становить 4 мг/кг (максимум 300 мг) кожні чотири тижні шляхом підшкірної ін'єкції. Рішення про подальше лікування препаратом Іларіс пацієнтів без клінічних покращень приймає лікар.
Подагричний артрит
Лікування слід проводити під наглядом лікарів із досвідом у діагностиці та лікуванні подагричного артриту і застосуванні біопрепаратів. Іларіс повинен вводити працівник охорони здоров'я.
Необхідний контроль гіперурикемії з відповідною терапією зниження рівня уратів. Іларіс потрібно застосовувати як терапію при потребі для лікування подагричних артритів.
Рекомендована доза препарату Іларіс для дорослих пацієнтів з подагричним артритом становить 150 мг підшкірно у вигляді разової дози під час нападу. Для досягнення максимального ефекту Іларіс слід застосовувати якомога швидше після початку нападу подагричного артриту.
Пацієнтам, у яких був відсутній ефект на початковій стадії лікування, не слід повторно застосовувати Іларіс. Для пацієнтів, у яких спостерігався ефект і для яких необхідне повторне лікування, інтервал між введенням доз повинен бути не менше ніж 12 тижнів.
Особливі популяції
Діти
Періодичні синдроми, пов'язані з кріопірином
Безпека та ефективність застосування препарату Іларіс
у пацієнтів із періодичними синдромами, пов'язаними з
кріопірином віком до 2 років не встановлені. Наявні на
сьогодні дані описані
в розділах «Фармакокінетика», «Фармакодинаміка», «Побічні
реакції», але ніяких рекомендацій щодо дозування не може
бути зроблено.
Системний ювенільний ідіопатичний артрит
Безпека та ефективність застосування препарату Іларіс для пацієнтів із періодичними синдромами, пов'язаними з кріопірином, віком до 2 років не встановлені.
Подагричний артрит
Досвіду застосування препарату Іларіс у дітей за показанням подагричний артрит немає.
Пацієнти літнього віку
Корекція дози не потрібна.
Немає значних відмінностей у профілі безпеки, що
спостерігається у пацієнтів віком понад
65 років.
Печінкова недостатність
Дані щодо застосування препарату Іларіс пацієнтам із порушенням функції печінки відсутні.
Ниркова недостатність
Для пацієнтів з нирковою недостатністю корекція дози не потрібна. Однак клінічний досвід застосування препарату таким пацієнтам обмежений.
Спосіб застосування
Іларіс 150 мг, порошок для розчину для ін'єкцій, постачається у флаконі одноразового використання для індивідуального застосування. Невикористаний продукт або відходи слід утилізувати відповідно до місцевих вимог.
Інструкції для розчинення
Використовуючи асептичну техніку, розвести вміст флакона при кімнатній температурі: повільно ввести 1,0 мл води для ін'єкцій за допомогою шприца 1 мл та голки 18 G × 50 мм. Обертати флакон повільно, нахиливши під кутом близько 45° протягом приблизно 1 хвилини, і залишити на 5 хвилин. Потім обережно повернути флакон догори дном і назад десять разів. По можливості не торкатися пальцями гумової пробки. Залишити на 15 хвилин при кімнатній температурі для отримання прозорого або опалесцентного розчину. Не струшувати. Не слід використовувати, якщо в розчині присутні частки.
Постукати по стінці флакона, щоб видалити залишки
рідини з пробки. Розчин має бути вільним від видимих
частинок, прозорим або опалесцентним. Розчин повинен бути
безбарвним, але може мати незначний коричнювато-жовтий
відтінок. Якщо розчин має яскраво коричневий колір, його не
слід застосовувати. Якщо розчин не був використаний відразу
ж після розчинення, то його слід зберігати при температурі
від 2 °С до 8 °С і використати протягом
24 годин.
Інструкції для застосування
Обережно наповнити шприц необхідною кількістю
розчину, залежно від дози (від 0,2 мл до
1,0 мл), та ввести підшкірно за допомогою голки 27 G × 13
мм.
Місця для ін'єкцій: верхня частина стегна, живіт, плече або сідниці. Слід уникати ділянки, де є ушкоджена шкіра, синці або висипи. Введення в рубцеві тканини слід уникати, оскільки це може знизити вплив препарату Іларіс.
Утилізація
Пацієнти або їхні опікуни повинні бути проінструктовані щодо утилізації флаконів, шприців та голок відповідно до місцевих вимог.
Діти.
У дослідження були включені 80 педіатричних пацієнтів віком 2-17 років із періодичними синдромами, пов'язаними з кріопірином. В цілому, не було жодних клінічно значущих відмінностей щодо безпеки та профілю толерантності Іларісу у педіатричних пацієнтів у порівнянні із загальною популяцією пацієнтів із періодичними синдромами, пов'язаними з кріопірином (що складалася з дорослих та педіатричних пацієнтів, N =3D 211), у тому числі щодо загальної частоти і тяжкості інфекційних епізодів. Інфекції верхніх дихальних шляхів були найбільш частими інфекційними захворюваннями.
Крім того, 6 педіатричних пацієнтів віком до 2 років були оцінені в невеликому відкритому клінічному дослідженні. Профіль безпеки препарату Іларіс схожий на такий у пацієнтів віком від 2 років.
Передозування
Інформація щодо передозування практично відсутня. Під час досліджень пацієнти та здорові добровольці отримували дози до 10 мг/кг внутрішньовенно або підшкірно без жодних ознак гострої токсичної дії препарату.
У разі передозування рекомендується проводити моніторинг стану пацієнтів і при необхідності негайно розпочати відповідну симптоматичну терапію.
Побічні реакції
До сліпих відкритих клінічних досліджень було залучено близько 2300 пацієнтів, у тому числі близько 250 дітей (віком від 2 до 17 років) з діагнозом періодичні синдроми, пов'язані з кріопірином, системний ювенільний ідіопатичний артрит, подагричний артрит або інші опосередковані ІL-1 бета захворювання, а також здорових добровольців. Найбільш частими побічними реакціями були інфекції (наприклад інфекції верхніх дихальних шляхів). Більшість реакцій були легкої або помірної тяжкості. Довгострокове лікування не мало впливу на тип або частоту побічних реакцій.
У пацієнтів, які застосовували Іларіс, спостерігалися випадки реакцій гіперчутливості.
При лікуванні препаратом Іларіс відмічались опортуністичні інфекції.
Періодичні синдроми, пов'язані з кріопірином
До клінічних досліджень було залучено 211 дорослих пацієнтів та пацієнтів дитячого віку (з діагнозами: сімейний холодовий аутозапальний синдром/ сімейна холодова кропив'янка, синдром Макла−Уельса та мультисистемне запальне захворювання неонатального віку/ хронічний дитячий неврологічний шкірно-артикулярний синдром). Безпека Іларісу порівнювалася з плацебо у базовому дослідженні фази ІІІ, що складалося з 8-тижневого відкритого періоду (частина 1), 24-тижневого рандомізованого подвійного сліпого та плацебо-контрольованого періоду виключення (частина 2) та 16-тижневого відкритого періоду із застосуванням Іларісу (частина 3). Усі пацієнти застосовували 150 мг препарату Іларіс підшкірно або 2 мг/кг маси тіла при вазі ≥ 15 кг та ≤ 40 кг.
Системний ювенільний ідіопатичний артрит
До клінічних досліджень Іларісу було залучено 201 пацієнта віком від 2 до 20 років із діагнозом системний ювенільний ідіопатичний артрит. Безпеку препарату Іларіс порівнювали із плацебо у двох пілотних дослідженнях ІІІ фази.
Подагричний артрит
До рандомізованих подвійних сліпих активних контрольованих клінічних досліджень тривалістю до 24 тижнів було залучено більше 700 пацієнтів з подагричним артритом, яким застосовували дози від 10 мг до 300 мг. Більше 250 пацієнтів пройшли лікування при рекомендованій дозі 150 мг у II і III фазі досліджень.
Побічні реакції перераховано відповідно до класів систем органів MedDRA і частоти. Категорії частоти визначаються таким чином: дуже часто (≥ 1/10); часто (≥ 1/100 до < 1/10); нечасто (≥ 1/1000 до < 1/100); рідко (≥ 1/10000 до < 1/1000), дуже рідко (< 1/10000), невідомо (частота не може бути оцінена за наявними даними). У кожній групі за частотою побічні реакції представлені в порядку зниження серйозності.
Таблиця 4. Побічні реакції, які спостерігалися у базовому дослідженні за участю пацієнтів із періодичними синдромами, пов'язаними з кріопірином, системним ювенільним ідіопатичним артритом, подагричним артритом
|
Класи систем органів |
Періодичні синдроми, пов'язані з кріопірином |
Системний ювенільний ідіопатичний артрит |
Подагричний артрит |
|
Інфекції та інвазії |
|||
|
Дуже часто |
Назофарингіт |
Пневмонія, гастроентерит, інфекції сечових шляхів, вірусні інфекції, синусити, риніти, фарингіти, тонзиліти, назофарингіти, інфекції верхніх дихальних шляхів |
Пневмонія, бронхіти, гастроентерити, інфекції сечових шляхів, грип, запалення пухкої клітковини, синусити, інфекції вуха, фарингіти, назофарингіти, інфекції верхніх дихальних шляхів |
|
Часто |
Інфекції сечових шляхів, інфекції верхніх дихальних шляхів, вірусні інфекції |
||
|
Порушення з боку нервової системи |
|||
|
Часто |
Запаморочення/вертиго |
Запаморочення/вертиго |
|
|
Порушення з боку травної системи |
|||
|
Дуже часто |
Біль у животі (верхня частина) |
||
|
Нечасто |
Гастроезофагеальна рефлюксна хвороба |
||
|
Порушення з боку шкіри та підшкірних тканин |
|||
|
Дуже часто |
Реакції у місці ін'єкції |
Реакції у місці ін'єкції |
|
|
Часто |
Реакції у місці ін'єкції |
||
|
Порушення з боку скелетно-м'язової системи та сполучної тканини |
|||
|
Дуже часто |
Артралгія |
||
|
Часто |
Скелетно-м'язовий біль |
Біль у спині |
|
|
Загальні порушення |
|||
|
Часто |
Втома/астенія |
||
|
Дослідження |
|||
|
Дуже часто |
Зниження рівня ниркового кліренсу креатиніну* Протеїнурія# Лейкопенія |
||
|
Часто |
Нейтропенія |
||
|
* Відповідно до оціненого кліренсу креатиніну більшість випадків були транзиторними. # Більшість випадків були представлені або транзиторними слідами, або реакцією на рівні 1+ на білок у сечі за методом тестових смужок. |
Дані довгострокових досліджень та лабораторні відхилення у пацієнтів із періодичними синдромами, пов'язаними з кріопірином
У ході клінічних випробувань Іларісу у пацієнтів із періодичними синдромами, пов'язаними з кріопірином, середні значення гемоглобіну підвищувалися, а рівні для білих клітин крові, нейтрофілів і тромбоцитів зменшувалися.
Підвищення рівня трансаміназ спостерігалося рідко.
Безсимптомні та помірні підвищення рівня сироваткового білірубіну без супутнього підвищення трансаміназ були відзначені у пацієнтів із періодичними синдромами, пов'язаними з кріопірином, які застосовували Іларіс.
У довгострокових, відкритих дослідженнях із збільшенням дози випадки інфекцій (гастроентерит, інфекції дихальних шляхів, інфекції верхніх дихальних шляхів), блювання і запаморочення були більш частими у групі дози 600 мг або 8 мг/кг, ніж в інших дозових групах.
Лабораторні відхилення у пацієнтів із системним ювенільним ідіопатичним артритом
Гематологія
В рамках загальної програми лікування системного
ювенільного ідіопатичного артриту минущі зниження рівня
білих клітин крові ≤ 0.8 x НМН були відзначені у 33
пацієнтів (16.5%). Минущі зниження абсолютного числа
нейтрофілів (АЧН) до рівня менше 1x 109/л
спостерігались
у 12 пацієнтів (6.0%). Минущі зниження кількості
тромбоцитів (< НМН) відмічались
у 19 пацієнтів (9.5%).
АЛТ/АСТ
В рамках загальної програми лікування системного ювенільного ідіопатичного артриту високі рівні АЛТ та/або АСТ > 3 x верхньої межі норми (ВМН) спостерігались у 19 пацієнтів (9.5%).
Лабораторні відхилення у пацієнтів із подагричним артритом
Гематологія
Зниження рівня білих клітин крові ≤ 0,8 × НМН (нижня межа норми) були зареєстровані у 6,7 % пацієнтів, які застосовували Іларіс, у порівнянні з 1,4 % пацієнтів, яких лікували триамцинолону ацетонідом. Зниження абсолютного числа нейтрофілів (АЧН) до рівня менше 1 × 109/л були відзначені у 2 % пацієнтів у порівняльних випробуваннях. Також спостерігалися поодинокі випадки виявлення рівня АЧН < 0,5 × 109/л.
Помірне (< НМН та > 75 × 109/л ) і
транзиторне зниження кількості тромбоцитів спостерігалося
із більш високою частотою (12,7 %) після застосування
Іларісу у активно контрольованих клінічних дослідженнях у
порівнянні із препаратом порівняння (7,7 %) у пацієнтів
із подагричним артритом.
Сечова кислота
Підвищення рівня сечової кислоти (0,7 мг/дл через 12
тижнів і 0,5 мг/дл через 24 тижні) спостерігалося після
лікування Іларісом в порівняльних випробуваннях у пацієнтів
з подагричним артритом. В іншому дослідженні серед
пацієнтів, які проходили ULT, збільшення рівня сечової
кислоти не спостерігалося. Збільшення рівня сечової кислоти
не спостерігалося у клінічних випробуваннях у групі
пацієнтів без подагричного артриту.
АЛТ/АСТ
Середні та медіанні збільшення аланінтрансамінази
(АЛТ) до 3,0 од/л і 2,0 од/л відповідно і
аспартаттрансамінази (АСТ) до 2,7 од/л і 2,0 од/л
відповідно в порівнянні
з вихідним рівнем до кінця дослідження спостерігалися в
групах Іларісу в порівнянні
з групою триамцинолону ацетоніду, однак частота клінічно
значущих змін (≥ 3 × ВМН) була більше у пацієнтів, які
отримували триамцинолону ацетонід (2,5 % для АСТ
та АЛТ) порівняно із групою лікування препаратом Іларіс
(1,6 % для АЛТ і 0,8 %
для АСТ).
Тригліцериди
В активних контрольованих клінічних випробуваннях за участю пацієнтів з подагричним артритом середнє збільшення рівня тригліцеридів становило 33,5 мг/дл в групі лікування пацієнтів препаратом Іларіс у порівнянні з помірним зниженням −3,1 мг/дл у групі триамцинолону ацетоніду. Частота випадків серед пацієнтів з підвищенням рівня тригліцеридів > 5 × ВМН становила 2,4 % для Іларісу і 0,7 % для триамцинолону ацетоніду. Клінічне значення цього спостереження невідоме.
Термін придатності. 3 роки.
Умови зберігання.
Зберігати при температурі 2-8 °C в оригінальній упаковці для захисту від світла. Не заморожувати. Зберігати у недоступному для дітей місці.
Несумісність
Через відсутність досліджень на сумісність цей лікарський засіб не слід змішувати з іншими лікарськими засобами.
Упаковка
По 150 мг порошку для розчину для ін'єкцій у 6 мл флаконі із безбарвного скла; по 1 флакону у коробці. Або по 4 коробки, кожна з яких містить 150 мг порошку для розчину для ін'єкцій у 6 мл флаконі із безбарвного скла, у пачці.
Категорія відпуску. За рецептом.
Виробник. Новартіс Фарма Штейн АГ.
Місцезнаходження виробника та його адреса місця провадження діяльності. Шаффхаусерштрассе, 4332 Штейн, Швейцарія.
Інші медикаменти цього ж виробника
Форма: порошок для суспензії для ін'єкцій по 30 мг, 1 флакон з мікросферами у комплекті з розчинником (натрію кармелозу/натрію карбоксиметилцелюлозу, маніт (Е 421), воду для ін'єкцій, полоксамер 188) по 2 мл у попередньо заповненому шприці, та одною голкою та одним адаптером в картонній коробці
Форма: таблетки, що диспергуються, по 3 мг № 30 (10х3) у блістерах
Форма: порошок для розчину для ін'єкцій по 75 мг; 1 флакон з порошком у комплекті з розчинником (вода для ін'єкцій) по 2 мл в ампулах № 1 в упаковці
Форма: розчин для ін'єкцій по 0,3 мг/1 мл, по 1 мл у ампулі; по 6 ампул в коробці; по 5 або по 10 коробок у пачці з картону
Форма: капсули м'які по 100 мг по 5 капсул у блістері; по 10 блістерів у картонній коробці