Соматулін Аутожель 90 Мг
Реєстраційний номер: UA/13432/01/03
- Соматулін Аутожель 120 мг
- Склад
- Фармакологічні властивості
- Клінічні характеристики
- Показання
- Протипоказання
- Особливості застосування
- Спосіб застосування та дози
- Початок лікування
- Адаптація лікування
- Спосіб введення лікарського засобу
- Діти
- Передозування
- Побічні реакції
- Клас систем
- органів
- Дуже часті (≥1/10)
- Часті (≥1/100 до <1/10)
- Нечасті (≥1/1000 до <1/100)
- Термін придатності
- Умови зберігання
- Упаковка
- Категорія відпуску
- Заявник
- Місцезнаходження заявника
Імпортер: ІПСЕН ФАРМА
Країна: ФранціяАдреса імпортера: 65, набережна Жорж Горс - 92100 Булонь Бійанкур, Франція
Форма
розчин для ін’єкцій пролонгованого вивільнення, 90 мг/шприц по 1 попередньо наповненому шприці для одноразового використання місткістю 0,5 мл з автоматичою захисною системою, 1 голкою (1,2х20 мм) в захисному ковпачку, у багатошаровому пакетику в картонній коробці
Склад
1 попередньо наповнений шприц містить ланреотид (у вигляді ланреотиду ацетату) 90 мг
Виробники препарату «Соматулін Аутожель 90 Мг»
Країна: Франція
Адреса: Парк д’актівіте дю Плато де Сінь департаментська дорога № 402, 83870 СІНЬ, Франція
Інструкція по застосуванню
для медичного застосування лікарського засобу
Соматулін Аутожель 60 мг
Соматулін Аутожель 90 мг
Соматулін Аутожель 120 мг
Склад
діюча речовина: lanreotide;
1 попередньо наповнений шприц містить ланреотид (у вигляді ланреотиду ацетату)
60 мг або 90 мг або 120 мг;
допоміжні речовини: кислота оцтова льодяна, вода для ін'єкцій.
Лікарська форма. Розчин для ін'єкцій пролонгованого вивільнення. Для глибокого підшкірного введення.
Основні фізико-хімічні властивості: гелеподібний розчин від білого до блідо-жовтого кольору.
Фармакотерапевтична група.
Гормони, що уповільнюють ріст. Код АТХ H01C B03.
Фармакологічні властивості
Фармакодинаміка.
Ланреотид є октапептидом, аналогом природного соматостатину. Подібно до природного соматостатину, ланреотид пригнічує ряд ендокринних, нейроендокринних, екзокринних та паракринних механізмів. Виявлено виражену тропність ланреотиду до соматостатинових рецепторів людини SSTR 2 та 5, низька тропність до SSTR 1, 3 та 4. Вважається, що активація соматостатинових рецепторів людини SSTR 2 та 5 є основним механізмом, що лежить в основі пригнічення секреції гормону росту (GH). Ланреотид більш активний, ніж природний соматостатин, та має більш тривалу дію. При цьому виражена селективність відносно гормону росту порівняно з інсуліном дає змогу застосовувати лікарський засіб у лікуванні акромегалії.
Ланреотид, подібно до соматостатину, чинить загальну екзокринну антисекреторну дію. Він пригнічує базальну секрецію мотиліну, шлункових інгібуючих пептидів та поліпептидів підшлункової залози, але не чинить значної дії на секрецію травних ферментів або шлункову секрецію. Окрім того, він знижує рівень плазмового хромограніну А та 5-HIAA (5-гідроксиіндолоцтової кислоти) сечової кислоти у пацієнтів із GEP-NETs і підвищеним рівнем цих пухлинних маркерів. Ланреотид значно пригнічує пов'язане з прийомом їжі посилення кровообігу у верхній брижовій артерії та ворітній вені. Ланреотид значно зменшує гідроелектролітичну секрецію в порожній кишці (секрецію води, натрію, калію та хлоридів), яка стимулюється простагландином Е1. Ланреотид знижує рівень пролактину у пацієнтів із акромегалією, які отримують лікування протягом тривалого часу. Ланреотид чинить пригнічувальну дію на екзокринну кишкову секрецію, травні гормони та механізм утворення кліткових протофібрил при його застосуванні в терапії симптомів ендокринних пухлин шлунково-кишкового тракту, особливо карциноїдних пухлин.
Було проведено 96-тижневе рандомізоване, подвійне сліпе, багатоцентрове дослідження третьої фази фіксованої тривалості із застосуванням лікарського засобу Соматулін Аутожель пацієнтам із нейроендокринними пухлинами шлунково-кишкового тракту або підшлункової залози для оцінювання антипроліферативної дії ланреотиду. Пацієнти були рандомізовані 1:1 для отримання лікарського засобу Соматулін Аутожель 120 мг кожні 28 днів (n=3D101) або ж плацебо (n=3D103). Рандомізація була стратифікована за попередньою терапією та наявністю/відсутністю прогресування на вихідному рівні за оцінкою RECIST 1.0 (Критерії оцінювання відповіді при солідних пухлинах) протягом скринінгової фази тривалістю 3-6 місяців. Пацієнти страждали на неоперабельні метастатичні та/або місцевопоширені захворювання з гістологічно підтвердженими добре або помірно диференційованими пухлинами з первинною локалізацією у підшлунковій залозі (44,6% пацієнтів), середній кишці (35,8% пацієнтів), задній кишці (6,9%) або ж із іншою/невідомою первинною локалізацією (12,7%). 69% пацієнтів із GEP-NETs мали пухлини 1-го ступеня диференціювання (G1), що визначалися за індексом проліферації Ki67 ≤ 2% (50,5% від загальної популяції пацієнтів) або ж мітотичним індексом <2 мітози/10 у полі зору (18,5% від загальної популяції пацієнтів), а 30% пацієнтів із GEP-NET мали пухлини в нижньому діапазоні 2-го ступеня (G2), що визначалися за індексом Ki67>2%-≤10%. У 1% пацієнтів відомості про ступінь були недоступні. З дослідження були виключені пацієнти з GEP-NET ступеня G2 із вищим індексом проліферації клітин (Ki 67>10%-≤20%) і нейроендокринними карциномами шлунково-кишкового тракту або підшлункової залози ступеня G3 (індекс Ki 67 > 20%).
Загалом, 52,5% пацієнтів мали печінкове пухлинне навантаження ≤10%, 14,5% мали печінкову пухлинну масу >10, а ≤25% і 33% мали печінкову пухлинну масу >25%.
Основною кінцевою точкою була виживаність без прогресування захворювання (PFS), вимірювана до прогресування захворювання за RECIST 1.0 або ж смерті протягом 96 тижнів після початку лікування. При аналізі PFS використовувалося незалежне централізоване радіологічне оцінювання прогресування захворювання.
Сприятливий вплив ланреотиду на зниження ризику прогресування або смерті був постійним, незалежно від локалізації первинної пухлини, печінкового пухлинного навантаження, попередньої хіміотерапії, вихідного значення Ki67, ступеня диференціювання пухлини або ж інших попередньо визначених характеристик. Клінічно значущий позитивний ефект від лікування лікарським засобом Соматулін Аутожель спостерігався у пацієнтів із пухлинами підшлункової залози, середньої кишки або іншого/невідомого походження, як у загальній популяції дослідження. Обмежена кількість пацієнтів із пухлинами задньої кишки (14 з 204) ускладнює інтерпретування результатів у цій підгрупі. На підставі наявних даних можна припустити відсутність ефекту ланреотиду в цих пацієнтів. Із плацебо на немаскований Соматулін Аутожель у розширеному дослідженні перейшли 45,6% (47 з 103) пацієнтів.
Фармакокінетика.
Фармакокінетичні параметри ланреотиду після внутрішньовенного введення здоровим добровольцям визначають обмежений позасудинний розподіл з рівноважним об'ємом розподілу 16,1 л. Загальний кліренс становив 23,7 л/год, період напіввиведення - 1,14 години, а середній час утримання - 0,68 години. При проведенні досліджень екскреції менше 5% ланреотиду потрапляли в сечу та менше 0,5% - у незміненому вигляді в кал, що свідчить про деяку біліарну екскрецію.
Після глибокого підшкірного введення Соматуліну Аутожелю 60 мг, 90 мг і 120 мг у здорових добровольців концентрація ланреотиду збільшується. Середня максимальна концентрація в сироватці крові становить 4,25, 8,39 і 6,79 нг/мл відповідно. Cmax досягається протягом першої доби після введення через 8, 12 і 7 годин відповідно (середнє значення). У середньому період напіввиведення становить 23,3, 27,4 і 30,1 дня відповідно. Через 4 тижні середня концентрація ланреотиду в сироватці становила 0,9, 1,11 і 1,69 нг/мл відповідно. Абсолютна біодоступність - 73,4, 69,0 і 78,4 % відповідно.
Після глибокого підшкірного введення Соматуліну Аутожелю 60 мг, 90 мг і 120 мг у пацієнтів з акромегалією значення середньої максимальної концентрації в сироватці крові 1,6, 3,5 і 3,1 нг/мл досягається протягом першої доби після введення через 6, 6 і 24 години відповідно з подальшим зниженням цього показника. Через 4 тижні після введення лікарського засобу середня концентрація ланреотиду в сироватці становила 0,7, 1,0 і 1,4 нг/мл відповідно.
Стійкий рівень ланреотиду досягається в середньому після 4 ін'єкцій кожні 4 тижні. В середньому після 4 ін'єкцій кожні 4 тижні середні значення Cmax становили 3,8, 5,7 і 7,7 нг/мл для дозувань 60 мг, 90 мг і 120 мг відповідно, середні значення Cmin становили 1,8, 2,5 і 3,8 нг/мл.
Лінійні фармакокінетичні профілі вивільнення спостерігалися після глибокого підшкірного введення лікарського засобу Соматулін Аутожель 60 мг, 90 мг і 120 мг у пацієнтів з акромегалією. Рівень ланреотиду у сироватці, отриманій після трьох глибоких підшкірних ін'єкцій лікарського засобу Соматулін Аутожель 60 мг, 90 мг або 120 мг кожні 28 днів, схожий з рівнем ланреотиду у сироватці у пацієнтів з акромегалію, які раніше отримували внутрішньом'язові ін'єкції Соматуліну 30 мг кожні 14, 10 і 7 днів відповідно.
У популяції фармакокінетичного аналізу у 290 пацієнтів з GEP-NET, які отримували Соматулін Аутожель у дозі 120 мг, спостерігалося швидке початкове вивільнення з середнім значенням Cmax 7,49 ± 7,58 нг/мл, що досягалося протягом першого дня після одноразового введення. Статична концентрація була досягнута після 5 ін'єкцій лікарського засобу Соматулін Аутожель 120 мг кожні 28 днів і підтримувалася до останньої оцінки (до 96 тижнів після першої ін'єкції). У стаціонарному стані середнє значення Cmax було 13,9 ± 7,44 нг/мл, а середній рівень в сироватці становив 6,56±1,99 нг/мл. Середній період напіврозпаду становив 49,8 ± 28,0 днів.
Ниркова/печінкова недостатність.
У ході дослідження у пацієнтів з гострою нирковою недостатністю спостерігається у середньому 2-кратне зниження загального сироваткового кліренсу з поступовим підвищенням періоду напіввиведення та AUC. У пацієнтів із помірною та гострою печінковою недостатністю спостерігалося 30-відсоткове зниження кліренсу. Об'єм розподілу та середній час утримання зросли у всіх дорослих учасників дослідження з печінковою недостатністю всіх стадій. Вплив на кліренс ланреотиду не спостерігався в популяції фармакокінетичного аналізу у пацієнтів з GEP-NET, включаючи 165 пацієнтів з легкою та помірною нирковою недостатністю (106 і 59 пацієнтів відповідно), які отримували Соматулін Аутожель. У пацієнтів з GEP-NET і тяжкими порушеннями функції нирок такі дані не вивчалися. У пацієнтів з GEP-NET та порушенням функції печінки (показники за Child-Pugh) такі дані не вивчалися. Немає необхідності змінювати початкову дозу для пацієнтів з нирковою та печінковою недостатністю, оскільки припускають, що концентрація ланреотиду в сироватці у такої групи пацієнтів буде в межах показників сироваткової концентрації, що безпечно переносяться здоровими добровольцями.
Літні пацієнти.
У літніх пацієнтів підвищується період напіввиведення та показник середнього утримання порівняно з такими у здорових молодих добровольців. Немає необхідності змінювати початкову дозу для літніх пацієнтів, оскільки припускають, що концентрація ланреотиду в сироватці у такої групи пацієнтів буде в межах показників сироваткової концентрації, що безпечно переносяться здоровими добровольцями.
У популяції фармакокінетичного аналізу у пацієнтів з GEP-NET, включаючи 122 пацієнти віком від 65 до 85 років, не спостерігалося ніякого впливу віку на кліренс та об'єм розподілу ланреотиду.
Клінічні характеристики
Показання
Лікування акромегалії при підвищеному рівні циркулюючого гормону росту (GH) та інсуліноподібного фактора росту (IGF-1) після оперативного втручання та/або радіотерапії або у разі, якщо протипоказані оперативне втручання та/або радіотерапія.
Лікування клінічних симптомів, спричинених акромегалією.
Лікування клінічних симптомів карциноїдних пухлин.
Лікування нейроендокринних пухлин шлунково-кишкового тракту або підшлункової залози (GEP-NETs) 1-го ступеня диференціювання та підмножини пухлин 2-го ступеня диференціювання (індекс Ki67 до 10%) походженням з середньої кишці, підшлункової залозі або з невідомим походженням, при виключенні походження з ділянок задньої кишки, у дорослих пацієнтів при нерезектабельних місцевопоширених або метастатичних пухлинах.
Протипоказання
Гіперчутливість до соматостатину або споріднених пептидів, а також до будь-якого з компонентів лікарського засобу.
Взаємодія з іншими лікарськими засобами та інші види взаємодій
Взаємодії, що передбачають дотримання запобіжних заходів у разі застосування.
Циклоспорин (перорально): зниження рівня циклоспорину в крові (зниження всмоктування циклоспорину в кишечнику). Збільшити дозу циклоспорину з проведенням контролю за рівнем циркуляції крові та знизити дозу після припинення лікування ланреотидом.
Інсулін, глітазони, репаглінід, препарати сульфонілсечовини: ризик гіпоглікемії та гіперглікемії: зниження потреби в лікуванні діабету після зниження або підвищення секреції ендогенного глюкагону. Необхідно посилити контроль за глікемією пацієнта, а дози лікарських засобів, які застосовують у терапії діабету, слід у разі необхідності коригувати під час лікування ланреотидом.
- Сумісне застосування лікарських засобів, індукуючих брадикардію (наприклад бета-блокаторів), може спричинити адитивний ефект на дещо знижений ритм серцевих скорочень, спричинених ланреотидом. Може бути необхідною корекція такого сумісного застосування лікарських засобів (див. розділ «Особливості застосування»).
- Обмежені опубліковані дані показують, що аналоги соматостатину можуть знижувати метаболічний кліренс сполук, які, як відомо, метаболізуються ферментами цитохрому Р450. Можливо, це відбувається через пригнічення гормону росту. Оскільки неможливо виключити те, що ланреотид може мати подібний ефект, інші лікарські засоби, які головним чином метаболізуються ферментом CYP3A4 та які мають невисокий терапевтичний індекс (наприклад хінідин), слід застосовувати з обережністю.
Інша інформація
Взаємодія з лікарськими засобами, які мають високий ступінь зв'язування з плазмою, є малоймовірною з огляду на помірну здатність ланреотиду до зв'язування з білками сироватки крові.
Особливості застосування
- Ланреотид може знижувати моторику жовчного міхура та спричиняти жовчнокам'яну хворобу. Тому може виникнути потреба у проведенні періодичного огляду. При тривалому лікуванні рекомендується проведення ехографії жовчного міхура перед лікуванням та кожні 6 місяців (див. розділ «Побічні реакції»).
- Ланреотид, подібно до соматостатину та інших його аналогів, пригнічує секрецію інсуліну та глюкагону. Тому у пацієнтів, які проходять лікування ланреотидом, можуть спостерігатися гіпоглікемія та гіперглікемія. Необхідно спостерігати за рівнем глюкози в крові на початку терапії ланреотидом або при зміні дози. Терапію діабету потрібно відповідно відкоригувати. Для пацієнтів, що хворіють на діабет та отримують інсулін, дозу інсуліну на початку знижують до 25 %, потім корегують згідно з рівнем глюкози в крові, який необхідно контролювати з початком лікування.
- Спостерігалось незначне зниження функції щитовидної залози при терапії ланреотидом у пацієнтів, хворих на акромегалію, хоча клінічний гіпотиреоз зустрічається рідко. При прояві симптоматики рекомендується дослідження функції щитовидної залози.
- У пацієнтів з акромегалією та первинною тиреотропною аденомою лікування ланреотидом повинно супроводжуватися контролем за об'ємом гіпофізарної пухлини.
- У пацієнтів, що не мають супутніх кардіологічних проблем, ланреотид може спричинити зниження частоти серцевих скорочень, хоча стан не межує неодмінно з брадикардією. У пацієнтів з уже наявними серцевими порушеннями може проявитися синусова брадикардія. Необхідно обережно починати лікування ланреотидом пацієнтам з брадикардією.
- Прояви значного та тривалого збільшення стеатореї передбачають комплементарне призначення панкреатичних екстрактів.
Застосування у період вагітності або годування груддю
Вагітність. Дослідження на тваринах не виявили будь-якої тератогенної дії, пов'язаної з ланреотидом, під час органогенезу. Кількість вагітних, які приймають ланреотид, дуже обмежена. Лікарський засіб слід призначати вагітним тільки у разі нагальної потреби.
Годування груддю. Невідомо, чи потрапляє даний лікарський засіб у грудне молоко. Оскільки багато лікарських засобів потрапляє в грудне молоко, необхідно прийняти рішення щодо припинення годування груддю або припинення застосування лікарського засобу, беручи до уваги необхідність лікування для матері.
Фертильність.
Зниження фертильності спостерігалось у самок щурів за рахунок інгібування секреції гормону росту при застосуванні в дозах, що перевищують терапевтичні дози для людини.
Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами
Жодного ефекту на здатність керувати автотранспортом чи працювати з іншими механізмами не встановлено, проте на фоні застосування лікарського засобу повідомляли про випадки запаморочення. Якщо у пацієнта спостерігається така реакція, слід утриматися від керування автотранспортом або роботи з іншими механізмами.
Спосіб застосування та дози
Соматулін Аутожель, розчин для ін'єкцій пролонгованої дії в попередньо наповненому шприці, представлений у трьох дозуваннях: 60 мг, 90 мг, 120 мг.
Акромегалія та лікування симптомів, спричинених карциноїдними пухлинами
Початок лікування
Акромегалія
На початку лікування рекомендована доза становить 60-120 мг кожні 28 днів.
Наприклад:
- у пацієнтів, що раніше отримували лікування Соматуліном 30 мг (порошок та розчинник для суспензії для ін'єкцій пролонгованого вивільнення (для внутрішньом'язових ін'єкцій)) кожні 14 днів, початкова доза Соматуліну Аутожелю повинна становити 60 мг кожні 28 днів;
- у пацієнтів, що раніше отримували лікування Соматуліном 30 мг (порошок та розчинник для суспензії для ін'єкцій пролонгованого вивільнення (для внутрішньом'язових ін'єкцій)) кожні 10 днів, початкова доза Соматуліну Аутожелю повинна становити 90 мг кожні 28 днів;
- у пацієнтів, що раніше отримували лікування Соматуліном 30 мг (порошок та розчинник для суспензії для ін'єкцій пролонгованого вивільнення (для внутрішньом'язових ін'єкцій)) кожні 7 днів, початкова доза Соматуліну Аутожелю повинна становити 120 мг кожні 28 днів.
Лікування симптомів, спричинених карциноїдними пухлинами
На початку лікування рекомендована доза становить 90 мг кожні 28 днів (4 тижні) протягом 2 місяців.
Адаптація лікування
Необхідно окремо коригувати терапію для кожного пацієнта в спеціалізованому відділенні. Дозу підбирають індивідуально, залежно від реакції пацієнта, яку оцінюють шляхом контролю за рівнем гормону росту (GH) та інсуліноподібного фактора росту (IGF-1) в плазмі, а також на підставі оцінки зміни клінічних симптомів.
Акромегалія
Рекомендується:
- зменшити дозу при нормалізації концентрацій (рівень гормону росту (GH) < 1 нг/мл та нормалізація рівня інсуліноподібного фактора росту (IGF-1) та/або зникнення клінічних симптомів);
- залишити дозу без змін, якщо рівень гормону росту (GH) знаходиться у межах від 2,5 нг/мл до 1 нг/мл;
- підвищити дозу при рівні гормону росту (GH) вище 2,5 нг/мл.
Пацієнтам, у яких на фоні лікування аналогами соматостатину було досягнуто ефективного контролю за захворюванням, можна призначити Соматулін Аутожель у дозі 120 мг кожні 42 або 56 днів.
Лікування симптомів, спричинених карциноїдними пухлинами
У разі недостатньої реакції, що оцінюється на підставі клінічних симптомів (припливи крові до обличчя та м'які випорожнення), дозу можна підвищити до 120 мг кожні 28 днів (4 тижні).
Якщо реакція достатня, що оцінюється на підставі клінічних симптомів (припливи крові до обличчя та м'які випорожнення), дозу можна знизити до 60 мг кожні 28 днів (4 тижні).
Лікування нейроендокринних пухлин шлунково-кишкового тракту або підшлункової залози (GEP-NETs) 1-го ступеня диференціювання та підмножини пухлин 2-го ступеня диференціювання (індекс Ki67 до 10%) походженням з середньої кишці, підшлункової залозі або з невідомим походженням, при виключенні походження з ділянок задньої кишки, у дорослих пацієнтів при нерезектабельних місцевопоширених або метастатичних пухлинах
Рекомендована доза становить одну ін'єкцію лікарського засобу Соматулін Аутожель 120 мг кожні 28 днів. Лікування лікарським Соматулін Аутожель слід продовжити стільки, скільки необхідно для контролю над пухлиною.
Пацієнти з порушенням функції нирок та печінки.
Зміна дози не потрібна (див. розділ «Фармакокінетика»).
Пацієнти літнього віку.
Зміна дози не потрібна (див. розділ «Фармакокінетика»).
Спосіб введення лікарського засобу
Розчин для ін'єкцій потрібно вводити глибоко підшкірно в верхній зовнішній квадрант сідниці або в верхню зовнішню ділянку стегна. Ін'єкції повинен проводити медичний працівник. У разі якщо пацієнти отримують стабільну дозу Соматуліну Аутожелю, ін'єкції може проводити сам пацієнт або його близькі після належного тренування, проведеного медичним працівником. При самостійному проведенні ін'єкції пацієнтом лікарський засіб необхідно вводити у верхню зовнішню ділянку стегна. Рішення щодо введення лікарського засобу пацієнтом чи особою, що пройшла тренування, приймає лікар. Незалежно від місця введення ін'єкції, шкіру не слід збирати в складку, а голку потрібно вводити швидко на всю її довжину перпендикулярно до поверхні шкіри. Необхідно вводити препарат по черзі в праву та ліву сідниці або стегна.
Інструкції із застосування
Соматулін Аутожель 60 мг, 90 мг, 120 мг, розчин для ін'єкцій пролонгованого вивільнення в попередньо наповненому шприці, призначений для негайного застосування після відкриття захисного пакетика. Важливо чітко дотримуватися інструкцій із застосування лікарського засобу. Не використовуйте лікарський засіб у разі пошкодження чи відкриття багатошарового пакетику. Невикористаний лікарський засіб та відходи необхідно утилізувати згідно з інструкціями, отриманими від медичного працівника.
Правила введення лікарського засобу
Лікарський засіб поставляється в готовому для використання попередньо наповненому шприці, оснащеному автоматичною системою захисту, що автоматично фіксується після введення лікарського засобу для запобігання уколу голкою після використання.
Рис. 1. Перед використанням:
Ковпачок (рис. 1а), захисний пристрій ходу поршня (рис. 1б).
Рис. 1а Рис. 1б

Рис. 2
Рис. 2. Після використання (голка в захисній системі).
При введенні лікарського засобу дотримуйтесь нижчезазначених рекомендацій.
1. Дістаньте Соматулін Аутожель з холодильника за 30 хвилин до проведення ін'єкції. Не відкривайте пакетик до проведення ін'єкції.
2. Перед тим як відкрити пакетик, переконайтеся, що він непошкоджений і термін придатності лікарського засобу не минув (рис. 3). Дата закінчення терміну придатності вказана на картонній коробці і на пакетику. Не застосовуйте лікарський засіб після закінчення терміну придатності або якщо упаковка пошкоджена.
Рис. 3
Примітка: mg (мг).
3. Вимийте руки з милом та переконайтеся, що місце для приготування ін'єкції є чистим.
4. Відкрийте пакетик і дістаньте попередньо наповнений шприц (рис. 4).
Рис. 4
5. Виберіть місце ін'єкції: верхній зовнішній квадрант сідниць (якщо ін'єкцію проводить медичний працівник або близька пацієнту людина) (рис. 5а) або верхня зовнішня частина стегна (якщо пацієнт самостійно вводить лікарський засіб) (рис. 5б).

Рис. 5а Рис. 5б
Чергуйте місце ін'єкції (правий та лівий бік) кожного разу, коли вводите лікарський засіб.
6. Продезінфікуйте місце передбачуваної ін'єкції, уникаючи розтирання шкіри.
7. Поверніть і зніміть захисний пристрій ходу поршня (рис. 6) та утилізуйте його.

Рис. 6
8. Видаліть ковпачок з голки (рис. 7) та утилізуйте його.
|
Рис. 7
9. Тримайте шкіру навколо місця ін'єкції великим і вказівним пальцями, див. рис. 8а (якщо ін'єкцію проводить медичний працівник або близька пацієнту людина) або рис. 8б (якщо пацієнт самостійно вводить лікарський засіб). Без натискання на шкіру в місці ін'єкції швидко введіть голку на всю довжину (глибокі підшкірні ін'єкції) перпендикулярно до шкіри (рис. 8в).
Рис. 8а або Рис. 8б
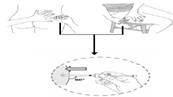
Рис. 8в
10. Вводьте лікарський засіб повільно (зазвичай необхідно 20 секунд) з рівномірним тиском на поршень до моменту, коли почуєте клацання. Продовжуйте утримувати поршень у натиснутому стані, щоб уникнути спрацювання автоматичної захисної системи (рис. 9).
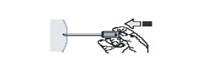
Рис. 9
11. Не відпускаючи поршень, витягніть голку з місця ін'єкції (рис. 10).

Рис. 10
12. Відпустіть поршень. Голка автоматично втягується в захисну систему, де вона буде заблокована назавжди (рис. 11).

Рис.11
13. Прикладіть сухий ватний тампон або стерильну марлю до місця ін'єкції, щоб запобігти кровотечі. Не слід розтирати та масажувати місце ін'єкції після введення.
14. Утилізуйте використаний шприц згідно з інструкцією, наданою лікарем або медичним працівником. Не викидайте пристрій в побутове сміття.
Діти
Лікарський засіб не рекомендується застосовувати дітям у зв'язку з відсутністю інформації щодо безпеки та ефективності для педіатричних пацієнтів.
Передозування
У разі передозування показана симптоматична терапія.
Побічні реакції
Під час проведення клінічних досліджень було зафіксовано побічні реакції у пацієнтів, що хворіють на акромегалію та GEP-NETs. Побічні реакції подано відповідно до такої класифікації: дуже часті (≥1/10); часті (≥1/100 до <1/10); нечасті (≥1/1000 до <1/100). Найбільш частими побічними реакціями після лікування ланреотидом є порушення з боку шлунково-кишкового тракту (найчастіше діарея, біль у животі, зазвичай легкі або помірні та тимчасового характеру), жовчнокам'яна хвороба (найчастіше безсимптомна) та реакції в місці введення (біль, вузлики, затвердіння). Профіль побічних реакцій аналогічний для всіх показань.
Клас системорганів |
Дуже часті (≥1/10) |
Часті (≥1/100 до <1/10) |
Нечасті (≥1/1000 до <1/100) |
Пост-реєстраційні дослідження з безпеки (частота невідома) |
|
Порушення з боку обміну речовин та харчування |
Гіпоглікемія, зниження апетиту**, гіперглікемія, цукровий діабет |
|||
|
Порушення з боку психіки |
Безсоння* |
|||
|
Порушення з боку нервової системи |
Запаморочення, головний біль, летаргія** |
|||
|
Порушення з боку серця |
Синусова брадикардія* |
|||
|
Порушення з боку судинної системи |
Припливи крові до обличчя* |
|||
|
Порушення з боку шлунково-кишкового тракту |
Діарея, рідке випорожнення*, біль у животі |
Нудота, блювання, запор, метеоризм, здуття живота, неприємні відчуття в животі, диспепсія, стеаторея** |
Зміна кольору калових мас* |
Панкреатит |
|
Порушення з боку гепатобіліарної системи |
Холелітіаз |
Розширення жовчних проток* |
||
|
Порушення з боку опорно-рухового апарату та сполучної тканини |
Скелетно м'язовий біль**, міалгія** |
|||
|
Порушення з боку шкіри та підшкірних тканин |
Алопеція, гіпотрихоз* |
|||
|
Загальні порушення та реакції в місці введення |
Астенія, стомлюваність, реакції в місці введення (біль, набряк, затвердіння, вузлики, свербіж) |
|||
|
Лабораторні показники |
Підвищення АЛТ*, відхилення від норми АСТ*, відхилення від норми АЛТ*, підвищення рівня білірубіну в крові*, підвищення рівня глюкози в крові*, підвищення рівня глікозильованого гемоглобіну*, зниження маси тіла*; зменшення рівня ферментів підшлункової залози** |
Підвищення АСТ*, підвищення рівня лужної фосфатази в крові*, відхилення від норми рівня білірубіну в крові*, зниження рівня натрію в крові* |
||
|
Порушення з боку імунної системи |
Алергічні реакції (у т.ч. ангіоневротичний набряк, анафілаксія, гіперчутливість) |
* За даними досліджень, проведених за участю пацієнтів з акромегалією.
** За даними досліджень, проведених за участю пацієнтів з GEP-NETs.
Повідомлення про підозрювані побічні реакції
Важливо повідомляти про підозрювані побічні реакції після реєстрації лікарського засобу. Це дає змогу здійснювати постійний моніторинг співвідношення користі та ризику застосування лікарського засобу. Медичні працівники повинні повідомляти про будь-які можливі побічні реакції через національну систему повідомлення.
Термін придатності
2 роки.
Умови зберігання
Зберігати у недоступному для дітей місці.
Зберігати при температурі від +2 до +8 °С в оригінальній упаковці. Не заморожувати. Для негайного використання після відкриття пакетику.
Упаковка
По 1 попередньо наповненому шприцу для одноразового використання місткістю
0,5 мл з автоматичною захисною системою, 1 голкою (1,2×20 мм) в захисному пластиковому ковпачку у багатошаровому пакетику в картонній коробці.
Категорія відпуску
За рецептом.
Виробник
ІПСЕН ФАРМА БІОТЕК (IPSEN PHARMA BIOTECH).
Місцезнаходження виробника та адреса місця провадження його діяльності
Парк д'aктівіте дю Плато де Сінь департаментська дорога № 402, 83870 СІНЬ, Франція/ Parc d'activites du Plateau de Signes chemin departemental № 402, 83870 SIGNES, France.
Заявник
ІПСЕН ФАРМА/IPSEN PHARMA.
Місцезнаходження заявника
65, набережна Жорж Горс-92100 Булонь Бійанкур, Франція/65, quai Georges Gorse-92100 Boulogne Billancourt, France).
Інші медикаменти цього ж виробника
Форма: суспензія оральна, 3 г по 10,27 г суспензії оральної в пакетику; по 12 пакетиків у картонній коробці
Форма: таблетки, вкриті плівковою оболонкою, по 20 мг № 28 (7х4) у блістерах, № 30 у пляшках
Форма: порошок для приготування розчину для перорального застосування по 10 г; по 10,167 г порошку в пакетику; по 10 або по 20 пакетів у картонній коробці
Форма: порошок для розчину для ін`єкцій, по 300 ОД; 1 флакон з порошком у картонній коробці
Форма: порошок по 0,1 мг та розчинник для розчину для ін'єкцій; по 7 флаконів з порошком та 7 ампул з 1 мл розчинника (розчин натрію хлориду 0,9 %) у картонній коробці