Мавенклад®
Реєстраційний номер: UA/17515/01/01
Імпортер: Арес Трейдінг С.А.
Країна: ШвейцаріяАдреса імпортера: Зон Індустрієль де Л'Урьєтаз, 1170 Обонн, Швейцарія
Форма
таблетки по 10 мг в алюмінієвому блістері № 1, № 4 або № 6 запечатаному у картонну обкладинку
Склад
1 таблетка містить 10 мг кладрибіну
Виробники препарату «Мавенклад®»
Країна: Італія
Адреса: Віале Пастер 10 (р-н Нервіано), 20014 Мілан (МІ), Італія
Країна: Італія
Адреса: Віа Б. Буоцци 2, 20090 Вімодроне (МІ), Італія
Інструкція по застосуванню
МАВЕНКЛАД®
(MAVENCLAD®)
Склад
діюча речовина: кладрибін;
1 таблетка містить 10 мг кладрибіну;
допоміжні речовини: гідроксипропілбетадекс, сорбіт (Е 420), магнію стеарат.
Лікарська форма. Таблетки.
Основні фізико-хімічні властивості: білі круглі двоопуклі таблетки діаметром 8,5 мм з тисненням «С» з одного боку та «10» − з іншого.
Фармакотерапевтична група. Селективні імуносупресанти. Кладрибін.
Код АТХ L04A A40.
Фармакологічні властивості.
Фармакодинаміка.
Механізм дії
Кладрибін є нуклеозидним аналогом дезоксіаденозину. Заміщення на атом хлору у пуриновому кільці захищає кладрибін від розкладу аденозиндезаміназою, збільшуючи час внутрішньоклітинного перебування проліків кладрибіну. Подальше фосфорилювання кладрибіну з утворенням його активної форми у вигляді трифосфату - 2-хлордезоксіаденозин-трифосфату (Cd‑ATP) - особливо ефективно досягається у лімфоцитах завдяки тому, що в них наявні постійно високі рівні дезоксицитидинкінази (DCK) та відносно низькі рівні 5'-нуклеотидази (5'-NTази). Високе співвідношення DCK до 5'-NTази сприяє кумуляції Cd‑ATP, що робить лімфоцити особливо чутливими до клітинної смерті. Внаслідок нижчого співвідношення DCK/5'-NTаза інші клітини, що походять зі спинного мозку, вражаються меншою мірою, ніж лімфоцити. DCK є ферментом, який обмежує швидкість перетворення кладрибіну в його активну трифосфатну форму, що призводить до селективного вичерпання Т- та В-клітин, які діляться та не діляться.
Основний механізм дії Cd‑ATP, що індукує апоптоз, має безпосередній та опосередкований вплив на синтез ДНК та функцію мітохондрій. У клітинах, що діляться, Cd‑ATP впливає на синтез ДНК шляхом інгібування рибонуклеотид-редуктази та конкурує з дезоксіаденозин-трифосфатом за вбудовування у ДНК за участю ДНК-полімераз. У клітинах, що знаходяться у стані спокою, кладрибін спричиняє одноланцюжковий розрив ДНК, швидке вичерпання нікотинамід-аденін-динуклеотиду, розщеплення АТФ та клітинну смерть. Існує доказ, що кладрибін також може спричиняти безпосередній, залежний та незалежний від каспази, апоптоз шляхом вивільнення цитохрому С та фактора, що індукує апоптоз, у цитозоль клітин, що не діляться.
Патогенез розсіяного склерозу (РС) залучає комплексний каскад явищ, у яких важливу роль відіграють різні типи імунних клітин, включаючи аутореактивні Т- та В-клітини. Механізм терапевтичної дії кладрибіну при РС ще повністю не вивчений, але вважається, що його основний вплив на В- та Т-лімфоцити перериває каскад імунних явищ, що мають вирішальне значення для РС.
Різні рівні експресії DCK та 5'-NTази у підтипах імунних клітин можуть пояснювати різницю у чутливості імунних клітин до кладрибіну. Завдяки цим рівням експресії клітини уродженої імунної системи менш вражаються, ніж клітини набутої імунної системи.
Фармакодинамічні ефекти
Було показано, що кладрибін проявляє тривалий ефект, переважно впливаючи на лімфоцити та аутоімунні процеси, залучені до патофізіології РС.
У дослідженнях найбільша частка пацієнтів з лімфопенією 3 або 4 ступеня (від < 500 до 200 клітин/мм³ або < 200 клітин/мм³) спостерігалась через 2 місяці після прийому першої дози кладрибіну кожного року, що вказує на наявність часового проміжку між перебуванням кладрибіну у плазмі та максимальним гематологічним ефектом.
Дані, одержані у клінічних дослідженнях для запропонованої кумулятивної дози 3,5 мг/кг маси тіла, вказують на поступове покращення медіанної кількості лімфоцитів до нормального діапазону на 84-му тижні після прийому першої дози кладрибіну (приблизно через 30 тижнів після прийому останньої дози кладрибіну). Кількість лімфоцитів у понад 75 % пацієнтів поверталась до нормального діапазону до 144-го тижня після прийому першої дози кладрибіну (приблизно через 90 тижнів після прийому останньої дози кладрибіну).
Лікування пероральним препаратом кладрибіну призводить до швидкого зменшення у кровотоці CD4+ та CD8+ Т-клітин. Для CD8+ T-клітин відбувається менш виражене зменшення і швидше відновлення, ніж для CD4+ T-клітин, що призводить до тимчасового зменшення співвідношення CD4 до CD8. Кладрибін зменшує кількість CD19+ B-клітин та клітин природних кілерів CD16+/CD56+, кількість яких також швидше відновлюється, ніж кількість CD4+ T-клітин.
Клінічна ефективність та безпека
РС з рецидивуюче-ремітуючим перебігом
Ефективність та безпека кладрибіну при пероральному застосуванні оцінювалися у рандомізованому подвійно-сліпому плацебо-контрольованому клінічному дослідженні (CLARITY) за участю 1326 пацієнтів з рецидивуюче-ремітуючим перебігом РС. Метою дослідження була оцінка ефективності кладрибіну порівняно з плацебо у зменшенні перерахованої на рік частоти рецидивів (ARR) (первинна кінцева точка), сповільненні прогресування інвалідизації та зменшенні активних уражень за даними МРТ.
Пацієнти приймали або плацебо (n =3D 437), або кумулятивну дозу кладрибіну 3,5 мг/кг (n =3D 433) чи 5,25 мг/кг маси тіла (n =3D 456) протягом 96-тижневого (2-річного) періоду досліджень, що складався з 2 лікувальних курсів. Пацієнтів у рандомізований спосіб розподіляли для прийому кумулятивної дози 3,5 мг/кг протягом першого курсу лікування на тижнях 1 та 5 першого року та другого курсу лікування на тижнях 1 та 5 другого року. Пацієнтів у рандомізований спосіб розподіляли для прийому кумулятивної дози 5,25 мг/кг протягом додаткового періоду лікування на тижнях 9 та 13 першого року. Більшість пацієнтів у групі плацебо (87,0 %) та групах лікування кладрибіном у дозах 3,5 мг/кг (91,9 %) та 5,25 мг/кг (89,0 %) завершили повний 96-тижневий період дослідження.
Пацієнти повинні були мати щонайменше 1 рецидив у попередні 12 місяців. У загальній досліджуваній популяції медіанний вік пацієнтів становив 39 років (діапазон від 18 до 65 років), а співвідношення жінок до чоловіків − приблизно 2 : 1. Середня тривалість захворювання на РС перед включенням у дослідження становила 8,7 року, а медіанний індекс базової неврологічної інвалідизації, встановлений в усіх групах лікування за розширеною шкалою оцінки ступеня інвалідизації Куртцке (EDSS), становив 3,0 (діапазон від 0 до 6,0). Понад дві третини пацієнтів до дослідження не приймали препаратів, що модифікують перебіг РС. Решта пацієнтів раніше лікувались інтерфероном бета-1а, інтерфероном бета-1b, глатирамеру ацетатом або наталізумабом.
У пацієнтів з рецидивуюче-ремітуючим перебігом РС, які приймали кладрибін у дозі 3,5 мг/кг, спостерігалось статистично значуще покращення порівняно з пацієнтами, які приймали плацебо, щодо перерахованої на рік частоти рецидивів, частки пацієнтів без рецидивів протягом 96 тижнів, частки пацієнтів без стійкої інвалідизації протягом 96 тижнів та часу до 3-місячного прогресування EDSS (див. таблицю 1).
|
Таблиця 1 Клінічні результати дослідження CLARITY (96 тижнів) |
|||
|
Параметр |
Плацебо
|
Кумулятивна доза кладрибіну |
|
|
3,5 мг/кг
|
5,25 мг/кг
|
||
|
Перерахована на рік частота рецидивів (95 % ДІ) |
0,33 (0,29; 0,38) |
0,14 (0,12; 0,17) p < 0,001* |
0,15 (0,12; 0,17) p < 0,001* |
|
- Відносне зменшення (кладрибін порівняно з плацебо) |
57,6 % |
54,5 % |
|
|
Частка пацієнтів без рецидивів протягом 96 тижнів |
60,9 % |
79,7 % |
78,9 % |
|
Час до 3-місячного прогресування EDSS, 10-й процентиль (місяців) |
10,8 |
13,6 |
13,6 |
|
- Співвідношення ризику (95 % ДІ) |
0,67 (0,48; 0,93) p =3D 0,018* |
0,69 (0,49; 0,96) p =3D 0,026* |
|
|
ДІ - довірчий інтервал * Порівняно з плацебо |
Результати лікування кладрибіном у дозі 3,5 мг/кг статистично значуще переважали плацебо щодо кількості та відносного зменшення T1 Gd+ уражень, активних T2 уражень та комбінованих неспецифічних уражень на томограмі мозку протягом повних 96 тижнів дослідження. Пацієнти, які приймали кладрибін, порівняно з групою плацебо мали відносне зменшення на 86 % середньої кількості уражень T1 Gd+ (відкоригована середня кількість для групи кладрибіну 3,5 мг/кг та групи плацебо становила 0,12 та 0,91 відповідно), відносне зменшення на 73 % середньої кількості активних T2 уражень (відкоригована середня кількість для групи кладрибіну 3,5 мг/кг та групи плацебо становила 0,38 та 1,43 відповідно) та відносне зменшення на 74 % середньої кількості комбінованих неспецифічних уражень в перерахунку на томограму пацієнта (відкоригована середня кількість для групи кладрибіну 3,5 мг/кг та групи плацебо становила 0,43 та 1,72 відповідно) (p < 0,001 за усіма 3 результатами МРТ).
Ретроспективний аналіз часу до 6-місячного підтвердженого прогресування EDSS виявив зменшення на 47 % ризику прогресування інвалідизації у групі, що приймала кладрибін у дозі 3,5 мг/кг, порівняно з групою плацебо (коефіцієнт ризику 0,53; 95 % ДІ [0,36; 0,79], p < 0,05).
Як показано у таблиці 1, вищі кумулятивні дози не надавали жодних клінічно значущих переваг, але були пов'язані з більшою частотою лімфопенії ступеня ≥ 3 (44,9 % у групі 5,25 мг/кг порівняно з 25,6 % у групі 3,5 мг/кг).
Пацієнти, які завершили участь у дослідженні CLARITY, могли бути включені до дослідження CLARITY Extension, первинною метою якого була оцінка безпеки. У цьому розширеному дослідженні 806 пацієнтів приймали або плацебо, або кумулятивну дозу кладрибіну 3,5 мг/кг (за режимом, подібним до того, який застосовувався у CLARITY) протягом періоду 96 тижнів.
Ефективність дії препарату щодо зменшення частоти рецидивів та уповільнення прогресування інвалідизації у пацієнтів, які приймали дозу 3,5 мг/кг протягом 2 років, підтримувалася у роки 3 та 4.
Ефективність лікування пацієнтів з високою активністю захворювання
Ретроспективно був проведений аналіз ефективності у підгрупах пацієнтів з високою активністю захворювання, які перорально приймали кладрибін у рекомендованій кумулятивній дозі 3,5 мг/кг. Ці підгрупи включали:
· пацієнтів з 1 рецидивом протягом попереднього року та щонайменше 1 ураженням T1 Gd+ або 9 чи більше ураженнями T2 під час терапії іншими препаратами, модифікуючими перебіг захворювання,
· пацієнтів з 2 або більше рецидивами протягом попереднього року незалежно від того, перебували вони на терапії препаратами, модифікуючими перебіг захворювання, чи ні.
Аналіз даних, одержаних у дослідженні CLARITY, показав подібний вплив лікування на частоту рецидивів, причому перерахована на рік частота рецидивів знаходилась у діапазоні від 0,16 до 0,18 у групах кладрибіну та від 0,47 до 0,50 у групі плацебо (p < 0,0001). Порівняно із загальною популяцією спостерігався більший вплив лікування на величину періоду до 6-місячної стійкої інвалідизації, коли кладрибін зменшував ризик прогресування інвалідизації на 82 % (коефіцієнт ризику 0,18; 95 % ДІ [0,07; 0,47]). Для плацебо 10-й процентиль для прогресування інвалідизації був досягнутий між 16 та 23 тижнями, тоді як у групах кладрибіну він не був досягнутий протягом усього терміну дослідження.
Рецидивуючий РС із вторинно-прогресуючим перебігом
Додаткове дослідження прийому кладрибіну додатково до інтерферону бета порівняно з прийомом плацебо та інтерферону бета також включало обмежену кількість пацієнтів із вторинно-прогресуючим перебігом РС (26 пацієнтів). У цих пацієнтів лікування кладрибіном у дозі 3,5 мг/кг призвело до зменшення перерахованої на рік частоти рецидивів порівняно з такою при застосуванні плацебо (0,03 порівняно з 0,30; співвідношення ризику: 0,11; p < 0,05). Між пацієнтами з рецидивуюче-ремітуючим РС та пацієнтами з рецидивуючим РС з вторинно-прогресуючим перебігом не спостерігалось різниці у перерахованій на рік частоті рецидивів. У жодній з цих підгруп не було показано впливу препарату на прогресування інвалідизації.
Пацієнти з вторинно-прогресуючим перебігом РС були виключені з дослідження CLARITY. Однак ретроспективний аналіз змішаних груп пацієнтів з досліджень CLARITY та ONWARD, яких визначали за базовим індексом EDSS ≥ 3,5 як індикатором РС з вторинно-прогресуючим перебігом, показав подібне зменшення перерахованої на рік частоти рецидивів порівняно з пацієнтами з індексом EDSS менше 3.
Фармакокінетика.
Кладрибін належить до проліків і має фосфорилюватися всередині клітини для того, щоб набути біологічної активності. Фармакокінетика кладрибіну досліджувалася після перорального та внутрішньовенного введення пацієнтам з РС та пацієнтам зі злоякісними новоутвореннями, а також на системах in vitro.
Абсорбція
Після перорального прийому кладрибін швидко абсорбується. Прийом 10 мг кладрибіну призводить до середньої Cmax кладрибіну в діапазоні від 22 до 29 нг/мл та відповідного середнього значення AUC в діапазоні від 80 до 101 нг×год/мл.
При пероральному прийомі кладрибіну натще медіанне значення Tmax становило 0,5 години (діапазон від 0,5 до 1,5 години). При прийомі з жирною їжею абсорбція кладрибіну відбувалась із затримкою (медіанне значення Tmax 1,5 години, діапазон від 1 до 3 годин) і Cmax зменшувалось на 29 %, тоді як значення AUC залишалось незміненим. Біодоступність пероральної дози 10 мг кладрибіну становила приблизно 40 %.
Розподіл
Кладрибін має великий об'єм розподілу, що вказує на його екстенсивний розподіл до тканин та внутрішньоклітинне поглинання. У дослідженнях було виявлено, що середній об'єм розподілу кладрибіну знаходиться в діапазоні від 480 до 490 л. Зв'язування з білками плазми становить 20 % і не залежить від концентрацій кладрибіну у плазмі.
Розподіл кладрибіну крізь біологічні мембрани здійснюється різними транспортними білками, включаючи ENT1, CNT3 та BCRP.
У дослідженнях іn vitro було показано, що ефлюкс кладрибіну лише мінімально пов'язаний з P-gp, тому клінічно значущі взаємодії з інгібіторами P-gp не очікуються.
Дослідження іn vitro показали незначуще потрапляння кладрибіну до гепатоцитів людини, опосередковане транспортерами.
Кладрибін має потенціал проникати крізь гематоенцефалічний бар'єр. Невелике дослідження за участю пацієнтів, що страждають на рак, показало, що співвідношення його концентрацій у спинномозковій рідині/плазмі становить приблизно 0,25.
Кладрибін та/або його фосфорильовані метаболіти значною мірою кумулюються та утримуються лімфоцитами людини. В умовах іn vitro було виявлено, що співвідношення внутрішньо- та зовнішньоклітинної кумуляції становить приблизно від 30 до 40 вже через 1 годину після початку експозиції.
Біотрансформація
Метаболізм кладрибіну вивчався у пацієнтів з РС після прийому однієї таблетки по 10 мг та разового внутрішньовенного введення дози 3 мг. Після як перорального, так і внутрішньовенного введення основним компонентом, який був виявлений у плазмі крові та сечі, була вихідна сполука кладрибіну. Вміст його метаболіту 2-хлораденіну як у плазмі крові, так і сечі був несуттєвим. У плазмі крові та сечі можна було знайти лише сліди інших метаболітів кладрибіну.
У печінкових системах in vitro спостерігався незначущий метаболізм кладрибіну (щонайменше 90 % складав незмінений кладрибін).
Кладрибін не є значущим субстратом для ферментів цитохрому Р450 і він не виявляє значного потенціалу як інгібітор CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 та CYP3A4. Не очікується, що інгібування цих ферментів або генетичний поліморфізм (наприклад CYP2D6, CYP2C9 або CYP2C19) призводитиме до клінічно значущого впливу на фармакокінетику або експозицію кладрибіну. Кладрибін не має клінічно значущого індуктивного впливу на ферменти CYP1A2, CYP2B6 та CYP3A4.
Після потрапляння до клітин-мішеней кладрибін фосфорилюється за участю DCK (а також за участю дезоксигуанозин-кінази в мітохондріях) до кладрибіну монофосфату (Cd-AMP), а далі − до кладрибіну дифосфату (Cd-ADP) та кладрибіну трифосфату (Cd-ATP). Дефосфорилювання та дезактивація Cd-AMP каталізується цитоплазматичною 5'-NTазою. У дослідженні внутрішньоклітинної фармакокінетики Cd-AMP та Cd-ATP за участю пацієнтів із хронічною мієлогенною лейкемією рівні Cd-ATP становили приблизно половину рівнів Cd-AMP.
Внутрішньоклітинний період напіввиведення становив 15 годин для Cd-AMP і 10 годин для Cd-ATP.
Виведення
На підставі об'єднаних популяційних фармакокінетичних даних, зібраних у різних дослідженнях, медіанні значення виведення становили 22,2 л/год для ниркового кліренсу та 23,4 л/год для нениркового кліренсу. Нирковий кліренс перевищував швидкість клубочкової фільтрації, що вказує на активну ниркову канальцеву секрецію кладрибіну.
Нениркова частина виведення кладрибіну (приблизно 50 %) включає незначущий печінковий метаболізм та екстенсивний внутрішньоклітинний розподіл і захоплення активного похідного кладрибіну (Cd‑ATP) цільовим внутрішньоклітинним компартментом (тобто лімфоцитами) з подальшим виведенням внутрішньоклітинного Cd-ATP згідно з життєвим циклом та шляхами виведення з цих клітин.
За оцінкою кінцевий період напіввиведення у типового пацієнта з популяції, для якої проводилась оцінка фармакокінетичних параметрів, становить приблизно 1 добу. Однак це не призводить до будь-якої кумуляції лікарського засобу після щоденного прийому дози, оскільки цей період напіввиведення стосується лише невеликої частки AUC.
Залежність від дози та часу
Після перорального прийому кладрибіну в дозах від 3 до 20 мг, Cmax та AUC зростали залежно від дози, що дає змогу припустити, що для пероральних доз до 20 мг на абсорбцію не впливають процеси, які обмежують її швидкість або ємність.
Після прийому повторних доз значної кумуляції кладрибіну в плазмі крові не спостерігалось. Немає вказівок на те, що фармакокінетика кладрибіну після повторного прийому може змінюватися залежно від часу.
Окремі групи пацієнтів
Оцінка фармакокінетики кладрибіну у пацієнтів з РС літнього або дитячого віку не проводилась, як і у пацієнтів з ураженням ниркової або печінкової функції.
Популяційний аналіз кінетики не показав жодного впливу віку (діапазон від 18 до 65 років) або статі пацієнта на фармакокінетику кладрибіну.
Ураження ниркової функції
Було показано, що нирковий кліренс кладрибіну залежить від кліренсу креатиніну. На підставі аналізу популяційної фармакокінетики, який включав дані пацієнтів з нормальною нирковою функцією та легким ураженням нирок, очікується, що загальний кліренс кладрибіну у пацієнтів з легким ураженням нирок (CLCR =3D 60 мл/хв) буде помірно зменшуватися, що призводитиме до зростання експозиції на 25 %.
Ураження печінкової функції
Роль печінкової функції у виведенні кладрибіну вважається незначущою.
Фармакокінетичні взаємодії
Дослідження лікарських взаємодій у пацієнтів з РС показало, що біодоступність 10 мг кладрибіну при пероральному прийомі не змінюється при одночасному застосуванні з пантопразолом.
Клінічні характеристики
Показання
Мавенклад® показаний для лікування дорослих пацієнтів з рецидивуючими формами розсіяного склерозу (РС) з високою активністю захворювання, встановленою на підставі клінічних або візуалізуючих обстежень.
Протипоказання
- Гіперчутливість до діючої речовини або до будь-яких допоміжних речовин препарату;
- інфекції з вірусом імунодефіциту людини (ВІЛ);
- активні хронічні інфекції (туберкульоз або гепатит);
- початок лікування кладрибіном пацієнтів з ослабленим імунітетом, включаючи пацієнтів, що приймають імуносупресуючу або мієлосупресуючу терапію;
- активні злоякісні новоутворення;
- помірне або тяжке ураження нирок (кліренс креатиніну < 60 мл/хв);
- період вагітності та годування груддю.
Взаємодія з іншими лікарськими засобами та інші види взаємодій
Мавенклад® містить гідроксипропілбетадекс, який може утворювати комплекси з іншими лікарськими засобами, що потенційно призводитиме до зростання біодоступності таких препаратів (особливо лікарських засобів з низькою розчинністю). Тому протягом обмеженої кількості днів прийому кладрибіну рекомендується приймати будь-які пероральні лікарські засоби щонайменше з 3-годинним інтервалом перед або після прийому Мавенкладу®.
Імуносупресуючі лікарські засоби
Пацієнтам з ослабленим імунітетом, включаючи пацієнтів, що проходять імуносупресуючу або мієлосупресуючу терапію, наприклад, метотрексатом, циклофосфамідом, циклоспорином або азатіоприном, або тим, хто постійно застосовує кортикостероїди, протипоказано розпочинати лікування кладрибіном через ризик додаткового впливу на імунну систему.
Під час лікування кладрибіном можна призначати інтенсивну короткотермінову терапію системними кортикостероїдами.
Інші лікарські засоби, що модифікують перебіг захворювання
Застосування Мавенкладу® з інтерфероном бета призводить до зростання ризику лімфопенії. Безпека та ефективність застосування Мавенкладу® в комбінації з іншими лікарськими засобами, що модифікують перебіг РС, не були встановлені, і тому таке одночасне лікування не рекомендується.
Гематотоксичні лікарські засоби
Оскільки кладрибін індукує зменшення кількості лімфоцитів, може очікуватися розвиток додаткових гематологічних побічних реакцій при прийомі кладрибіну перед або одночасно з іншими речовинами, що впливають на гематологічний профіль (такими як карбамазепін). У таких випадках рекомендується ретельний контроль гематологічних параметрів.
Живі або живі атенуйовані вакцини
Лікування Мавенкладом® не слід розпочинати в межах 4 − 6 тижнів після вакцинації живими або живими атенуйованими вакцинами через ризик гострого інфікування вакциною. Під час та після лікування кладрибіном слід уникати вакцинації живими або живими атенуйованими вакцинами доти, доки кількість лімфоцитів у пацієнта не повернеться в межі норми.
Сильні інгібітори транспортерів ENT1, CNT3 та BCRP
На рівні абсорбції кладрибіну єдиним імовірним джерелом взаємодій, що має клінічне значення, є білок резистентності раку молочної залози (BCRP або ABCG2). Інгібування BCRP у шлунково-кишковому тракті може збільшувати пероральну біодоступність та системну дію кладрибіну. Відомі інгібітори BCRP, які in vivo можуть змінювати фармакокінетику субстратів BCRP на 20 %, включають ельтромбопаг.
Дослідження in vitro вказують на те, що кладрибін є субстратом рівноважного нуклеозидного (ENT1) та концентруючого нуклеозидного (CNT3) транспортних білків. Відповідно, на біодоступність, внутрішньоклітинний розподіл та ниркове виведення кладрибіну теоретично можуть впливати сильні інгібітори транспортерів ENT1, CNT3 та BCRP, такі як дилазеп, ніфедипін, німодипін, цилостазол, суліндак та резерпін. Однак підсумковий вплив, що виражається у зміні потенційної дії кладрибіну, важко передбачити.
Хоча клінічне значення таких взаємодій невідоме, під час 4- або 5-денного лікування кладрибіном рекомендується уникати одночасного прийому сильних інгібіторів ENT1, CNT3 та BCRP. Якщо це неможливо, слід розглянути підбір альтернативних лікарських засобів для одночасного застосування, які не мають або мають мінімальні інгібуючі властивості відносно транспортерів ENT1, CNT3 та BCRP. Якщо це неможливо, рекомендується зменшити дозу лікарських засобів, що містять такі сполуки, до мінімальної обов'язкової дози, розділити прийом препаратів у часі та ретельно контролювати стан пацієнта.
Сильні індуктори транспортерів BCRP та P-gp
Вплив сильних індукторів ефлюксних транспортерів BCRP та P‑глікопротеїну (P‑gp) на біодоступність та розподіл кладрибіну у спеціальних дослідженнях не вивчався. При одночасному прийомі сильних індукторів транспортерів BCRP (наприклад кортикостероїдів) або P-gp (наприклад рифампіцину, звіробою продірявленого) слід враховувати можливе послаблення дії кладрибіну.
Гормональні контрацептиви
На сьогодні невідомо, чи може кладрибін зменшувати ефективність гормональних контрацептивів системної дії. Тому жінки, які застосовують гормональні контрацептиви системної дії, повинні додавати бар'єрні засоби контрацепції під час лікування кладрибіном та протягом щонайменше 4 тижнів після прийому останньої дози у кожний рік лікування (див. розділ «Застосування у період вагітності або годування груддю»).
Особливості застосування
Гематологічний моніторинг
Механізм дії кладрибіну тісно пов'язаний зі зменшенням кількості лімфоцитів. Вплив на кількість лімфоцитів є дозозалежним. У клінічних дослідженнях також спостерігалось зменшення кількості нейтрофілів, кількості еритроцитів, гематокриту, гемоглобіну та кількості тромбоцитів порівняно з базовими значеннями, хоча ці параметри зазвичай залишались у межах норми.
При прийомі кладрибіну перед або одночасно з іншими речовинами, які впливають на гематологічний профіль, можна очікувати розвитку додаткових побічних реакцій.
Кількість лімфоцитів необхідно визначати:
· перед початком прийому Мавенкладу® у рік 1,
· перед початком прийому Мавенкладу® у рік 2,
· через 2 та 6 місяців після початку лікування у кожному році лікування; якщо кількість лімфоцитів нижче 500 клітин/мм³, цей параметр слід активно контролювати доти, поки значення не збільшиться знову.
Щодо прийняття рішення стосовно лікування на підставі кількості лімфоцитів див. розділ «Спосіб застосування та дози» та підрозділ «Інфекції» нижче.
Інфекції
Кладрибін може послаблювати імунний захист організму, тим самим збільшуючи імовірність інфекцій. Інфекція ВІЛ, активний туберкульоз та активний гепатит мають бути виключені до початку лікування кладрибіном (див. розділ «Протипоказання»).
Під час лікування можуть активуватися латентні інфекції, включаючи туберкульоз або гепатит. Тому перед початком терапії у рік 1 та рік 2 необхідно провести перевірку щодо наявності латентних інфекцій, зокрема туберкульозу та гепатитів В і С. Початок лікування Мавенкладом® необхідно відкласти до адекватного виліковування інфекцій.
У пацієнтів з гострими інфекціями також слід врахувати можливість відтермінування початку лікування кладрибіном доти, поки інфекції не будуть повністю контрольованими.
Особливу увагу рекомендується приділяти пацієнтам, у яких немає історії контакту з вірусом вітряної віспи. Перед початком терапії кладрибіном рекомендується провести вакцинацію пацієнтів з відсутніми антитілами до вітряної віспи. У такому випадку початок лікування Мавенкладом® слід відкласти на 4 − 6 тижнів, щоб досягти повного ефекту від вакцинації.
У пацієнтів, що приймали кладрибін, зростала частота випадків оперізуючого герпесу. Якщо кількість лімфоцитів падає нижче 200 клітин/мм³, під час лімфопенії 4 ступеня слід врахувати проведення протигерпесної профілактики згідно з місцевими стандартними протоколами.
У пацієнтів з кількістю лімфоцитів нижче 500 клітин/мм³ слід проводити активний моніторинг ознак та симптомів, що дають змогу припустити наявність інфекції, зокрема оперізуючого герпесу. Якщо такі ознаки та симптоми спостерігаються, слід розпочати лікування інфекції за клінічними показаннями. Можна розглянути доцільність переривання або відтермінування лікування Мавенкладом® до належного виліковування інфекції.
Повідомлялось про випадки прогресуючої мультифокальної лейкоенцефалопатії (ПМЛ) при застосуванні парентеральних препаратів кладрибіну у пацієнтів, які лікували волосатоклітинний лейкоз за різними схемами лікування.
У базі даних клінічних досліджень застосування кладрибіну при РС (1976 пацієнтів, 8650 пацієнто-років) повідомлення про ПМЛ відсутні. Однак перед початком лікування Мавенкладом® (зазвичай в межах 3 місяців) слід провести базове МРТ-дослідження.
Злоякісні новоутворення
У клінічних дослідженнях випадки злоякісних новоутворень спостерігались частіше у пацієнтів, що приймали кладрибін, порівняно з пацієнтами, які приймали плацебо.
Мавенклад® протипоказаний пацієнтам з РС, що мають активні злоякісні новоутворення (див. розділ «Протипоказання»). У пацієнтів з попередніми злоякісними новоутвореннями перед початком лікування слід провести індивідуальну оцінку переваг ти ризиків застосування Мавенкладу®. Пацієнтам, що приймають Мавенклад®, слід порадити виконувати стандартні рекомендації щодо обстежень на рак.
Контрацепція
Перед початком лікування у рік 1 та рік 2 жінок репродуктивного віку та чоловіків, які потенційно можуть стати батьками, слід поінформувати про потенційний серйозний ризик для плода та необхідність вжиття ефективних заходів з контрацепції.
Жінки репродуктивного віку повинні запобігати настанню вагітності, застосовуючи ефективні засоби контрацепції під час лікування кладрибіном і щонайменше протягом 6 місяців після прийому останньої дози.
Пацієнти-чоловіки повинні вживати заходів для запобігання вагітності своєї партнерки під час лікування кладрибіном і щонайменше протягом 6 місяців після прийому останньої дози.
Переливання крові
Для пацієнтів, які потребують переливання крові, рекомендується проводити променеву обробку клітинних компонентів крові перед введенням для запобігання пов'язаних з переливанням захворювань «трансплантат проти хазяїна». Рекомендується проконсультуватися з лікарем-гематологом.
Перехід на та з лікування кладрибіном
Перед початком застосування Мавенкладу® для пацієнтів, які раніше приймали імуномодулюючі або імуносупресуючі лікарські засоби, слід врахувати механізм дії та тривалість ефекту цих препаратів. Потенційний додатковий вплив на імунну систему також слід враховувати, якщо такі лікарські засоби застосовуються після лікування Мавенкладом®.
При переході з іншого лікарського засобу для лікування РС рекомендується провести базове МРТ-сканування (див. підрозділ «Інфекції» вище).
Ураження печінки
Хоча важливість печінкової функції для виведення кладрибіну вважається незначущою, через відсутність даних застосування Мавенкладу® не рекомендується пацієнтам з помірним або тяжким ураженням печінки (індекс Чайлда−П'ю > 6).
Непереносимість фруктози
Мавенклад® містить сорбіт. Пацієнтам зі спадковими проблемами з переносимістю фруктози не слід приймати цей лікарський засіб.
Застосування у період вагітності або годування груддю.
Контрацепція у чоловіків та жінок
Перед початком лікування у рік 1 та рік 2 жінок репродуктивного віку та чоловіків, які потенційно можуть стати батьками, слід поінформувати про потенційний серйозний ризик для плода та необхідність вжиття ефективних заходів з контрацепції.
У жінок репродуктивного віку перед початком лікування Мавенкладом® у роки 1 та 2 потрібно виключити вагітність, а настанню вагітності слід запобігати, застосовуючи ефективні засоби контрацепції під час лікування кладрибіном та протягом щонайменше 6 місяців після прийому останньої дози. Жінки, які застосовують гормональні контрацептиви системної дії, повинні додавати бар'єрні засоби контрацепції під час лікування кладрибіном та протягом щонайменше 4 тижнів після прийому останньої дози у кожний рік лікування. Жінки, які завагітніли під час лікування Мавенкладом®, повинні припинити терапію.
Оскільки кладрибін впливає на синтез ДНК, слід очікувати його негативного впливу на гаметогенез людини. Тому пацієнти-чоловіки повинні вживати заходів, щоб запобігти настанню вагітності у своєї партнерки під час лікування кладрибіном та протягом щонайменше 6 місяців після прийому останньої дози.
Вагітність
З огляду на досвід застосування інших речовин, що інгібують синтез ДНК у людини, кладрибін може спричиняти уроджені вади розвитку плода у разі прийому у період вагітності. У дослідженнях на тваринах була показана репродуктивна токсичність кладрибіну.
Мавенклад® протипоказаний вагітним жінкам (див. розділ «Протипоказання»).
Годування груддю
Невідомо, чи виділяється кладрибін у грудне молоко людини. Через потенційні серйозні побічні реакції у немовлят, що знаходяться на грудному вигодовуванні, годування груддю протипоказане під час лікування Мавенкладом® і протягом 1 тижня після прийому останньої дози (див. розділ «Протипоказання»).
Фертильність
У мишей не спостерігалось впливу кладрибіну на фертильність або репродуктивну функцію потомства, однак у мишей та мавп спостерігався вплив на тестикулярну функцію.
Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами.
Мавенклад® не впливає або майже не впливає на здатність пацієнтів керувати автомобілем та працювати з іншими механізмами.
Спосіб застосування та дози
Лікування Мавенкладом® повинно розпочинатися та проводитися під наглядом лікаря, який має досвід лікування РС.
Дози
Рекомендована кумулятивна доза Мавенкладу® становить 3,5 мг/кг маси тіла протягом 2 років і призначається у вигляді 1 курсу лікування дозою 1,75 мг/кг щорічно. Кожний курс лікування складається з 2 тижнів лікування, один - на початку першого місяця, а інший - на початку другого місяця відповідного року лікування. Кожний лікувальний тиждень складається з 4 або 5 днів, в які пацієнт приймає 10 мг або 20 мг (одну або дві таблетки) у вигляді разової добової дози, залежно від маси тіла. Подальшу детальну інформацію наведено у таблицях 2 та 3.
Після завершення 2 курсів лікування подальшого лікування кладрибіном у роки 3 та 4 не потрібно. Повторний початок лікування через 4 роки не вивчався.
Критерії для початку та продовження терапії
Кількість лімфоцитів повинна бути:
· нормальною перед початком прийому Мавенкладу® у рік 1,
· щонайменше 800 клітин/мм³ перед початком прийому Мавенкладу® у рік 2.
У разі необхідності для відновлення кількості лімфоцитів курс лікування у рік 2 може бути відтермінований до 6 місяців. Якщо таке відновлення потребує більше 6 місяців, пацієнт більше ніколи не може приймати Мавенклад®.
Розподіл дози
Розподіл загальної дози протягом 2 років лікування наведено в таблиці 2. Для деяких пацієнтів, зважаючи на масу тіла, кількість таблеток може бути різною в перший та другий лікувальні тижні. Застосування пероральних форм кладрибіну пацієнтами з масою тіла менше 40 кг не вивчалось.
Таблиця 2 Доза Мавенкладу® залежно від маси тіла пацієнта у лікувальний тиждень кожного року лікування
|
Діапазон маси тіла, кг |
Доза у мг (кількість таблеток по 10 мг) у лікувальний тиждень |
|
|
Лікувальний тиждень 1 |
Лікувальний тиждень 2 |
|
|
від 40 до < 50 |
40 мг (4 таблетки) |
40 мг (4 таблетки) |
|
від 50 до < 60 |
50 мг (5 таблеток) |
50 мг (5 таблеток) |
|
від 60 до < 70 |
60 мг (6 таблеток) |
60 мг (6 таблеток) |
|
від 70 до < 80 |
70 мг (7 таблеток) |
70 мг (7 таблеток) |
|
від 80 до < 90 |
80 мг (8 таблеток) |
70 мг (7 таблеток) |
|
від 90 до < 100 |
90 мг (9 таблеток) |
80 мг (8 таблеток) |
|
від 100 до < 110 |
100 мг (10 таблеток) |
90 мг (9 таблеток) |
|
від 110 та більше |
100 мг (10 таблеток) |
100 мг (10 таблеток) |
У таблиці 3 показано, як загальну кількість таблеток на тиждень лікування розподілити по окремих днях. Рекомендується кожну добову дозу кладрибіну у кожному лікувальному тижні приймати з інтервалами 24 години, приблизно в один й той самий час дня. Якщо добова доза становить дві таблетки, обидві таблетки приймають разом як одну дозу.
|
Таблиця 3 Розподіл таблеток Мавенкладу® по 10 мг по дням тижня |
|||||
|
Загальна кількість таблеток на тиждень |
День 1 |
День 2 |
День 3 |
День 4 |
День 5 |
|
4 |
1 |
1 |
1 |
1 |
0 |
|
5 |
1 |
1 |
1 |
1 |
1 |
|
6 |
2 |
1 |
1 |
1 |
1 |
|
7 |
2 |
2 |
1 |
1 |
1 |
|
8 |
2 |
2 |
2 |
1 |
1 |
|
9 |
2 |
2 |
2 |
2 |
1 |
|
10 |
2 |
2 |
2 |
2 |
2 |
Пропущену дозу необхідно прийняти якомога скоріше після з'ясування пропуску того ж самого дня згідно з режимом лікування.
Пропущену дозу не можна приймати разом з наступною дозою за схемою лікування наступного дня. У разі пропущеної дози пацієнт повинен прийняти її наступного дня та збільшити кількість днів лікування у цей лікувальний тиждень. Якщо пропущено дві послідовні дози, застосовується такий же підхід і кількість днів у лікувальному тижні збільшується на два дні.
Одночасне застосування інших пероральних лікарських засобів
Протягом обмеженої кількості днів прийому кладрибіну рекомендується відокремлювати прийом будь-яких інших пероральних лікарських засобів від прийому Мавенкладу® щонайменше на 3 години.
Окремі групи пацієнтів
Пацієнти з ураженням нирок
Спеціальних досліджень лікування пацієнтів з ураженням нирок не проводилось.
Для пацієнтів з легким ураженням нирок (кліренс креатиніну від 60 до 89 мл/хв) корекція дози не вважається потрібною.
Безпека та ефективність застосування кладрибіну пацієнтами з помірним або тяжким ураженням нирок не вивчалися. Тому Мавенклад® протипоказаний таким пацієнтам (див. розділ «Протипоказання»).
Пацієнти з ураженням печінки
Досліджень лікування пацієнтів з ураженням печінки не проводилось.
Хоча важливість печінкової функції у виведенні кладрибіну вважається незначущою, через відсутність даних застосування Мавенкладу® не рекомендується пацієнтам з помірним або тяжким ураженням печінки (індекс Чайлда−П'ю > 6).
Пацієнти літнього віку
Клінічні дослідження перорального застосування кладрибіну при РС не включали пацієнтів віком понад 65 років, тому невідомо, чи будуть вони відповідати на лікування інакше, ніж молодші пацієнти.
Пацієнтам літнього віку Мавенклад® рекомендується застосовувати з обережністю, враховуючи потенційно більшу частоту випадків зниження печінкової або ниркової функції, супутні захворювання та інші види терапії, що проводяться.
Спосіб застосування
Мавенклад® призначений для перорального застосування. Таблетки слід приймати з водою та ковтати не розжовуючи. Таблетки можна приймати незалежно від прийому їжі.
Оскільки таблетки не вкриті оболонкою, їх потрібно ковтати негайно після виймання з блістера, не залишати на поверхні та не тримати у руках довше, ніж це потрібно для прийому дози. Якщо таблетка залишалась на поверхні або якщо розламану чи розкришену таблетку вийняли з блістера, слід ретельно вимити водою поверхню контакту з таблеткою.
Пацієнт має брати таблетки сухими руками, після чого руки слід ретельно вимити.
Інструкція для самостійного прийому пацієнтом таблеток Мавенклад®
- Приготуйте стакан з водою та впевніться, що у Вас сухі та чисті руки.
- Візьміть коробку з препаратом, повернувши її до себе стороною, на якій надруковані інструкції.
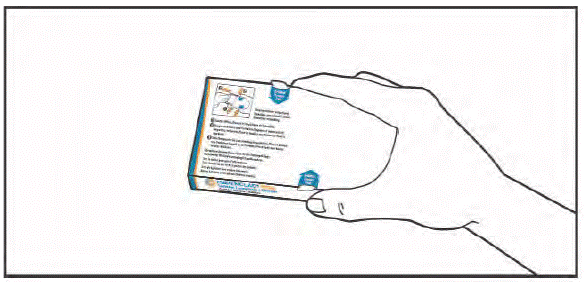
- (1) Відкрийте клапан з лівого боку коробки.
(2) Одночасно натисніть на гачки на коробці з обох сторін вказівним та великим пальцями та тримайте їх у такому положенні.
(3) Потягніть контурну чарункову упаковку, поки вона рухається. УВАГА: не виймайте контурну чарункову упаковку з коробки.
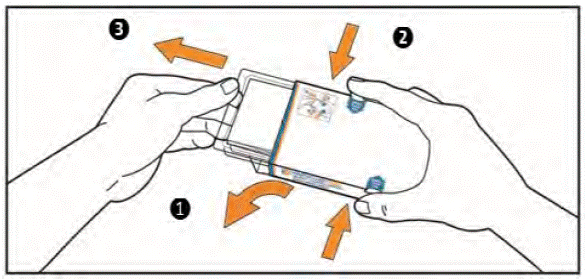
- Витягніть з коробки інструкцію для медичного застосування. Впевніться, що Ви уважно ознайомилися з її змістом, у тому числі з інструкцією для самостійного прийому таблеток.
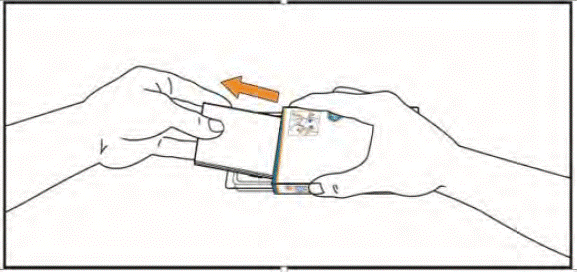
- Припідніміть блістер, натискаючи пальцями на отвір у контурній чарунковій упаковці. Помістіть руку під блістер та видавіть у руку 1 або 2 таблетки, як Вам призначив лікар.
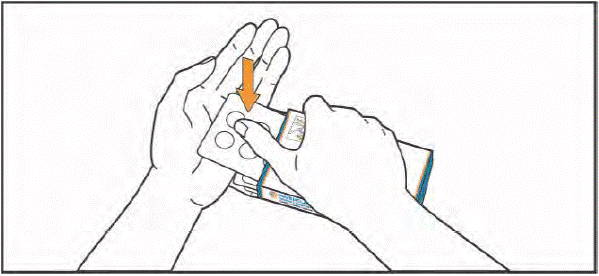
- Прийміть таблетки, запиваючи їх водою. Таблетки необхідно ковтати цілими, не розжовуючи та не дозволяючи їм розчинитися у роті. Контакт таблеток зі шкірою має бути обмежений. Не торкайтеся руками носа, очей та інших частин тіла.
- Після прийому таблеток ретельно вимийте руки водою з милом.
- Вкладіть контурну чарункову упаковку в коробку. Зберігайте її в оригінальній упаковці для захисту від вологи.
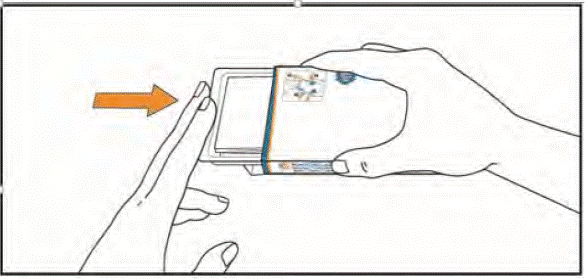
До прийому наступної дози зберігайте таблетки у блістері. Не видавлюйте таблетки з блістера та не зберігайте їх в іншому контейнері.
Діти.
Безпека та ефективність застосування Мавенкладу® пацієнтами віком до 18 років не були встановлені. Відповідні дані відсутні.
Передозування
Існує обмежений досвід передозування кладрибіну при пероральному прийомі. Відомо, що лімфопенія, яка розвивається внаслідок цього, є дозозалежною.
У пацієнтів з передозуванням кладрибіну рекомендується проведення особливо ретельного моніторингу гематологічних параметрів.
Специфічний антидот до кладрибіну невідомий. Лікування полягає у ретельному спостереженні за станом пацієнта та проведенні відповідних підтримуючих заходів. Слід врахувати можливу необхідність відміни Мавенкладу®. Через швидкий та екстенсивний внутрішньоклітинний та тканинний розподіл, малоймовірно, що кладрибін буде значною мірою виводитися при проведенні гемодіалізу.
Побічні реакції
Стислий опис профілю безпеки
Більшість клінічно значущих побічних реакцій, про які повідомлялось у пацієнтів з РС, що приймали кладрибін у рекомендованій кумулятивній дозі 3,5 мг/кг протягом 2 років у клінічних дослідженнях, були представлені лімфопенією та оперізуючим герпесом. Частота випадків оперізуючого герпесу була вищою протягом періоду, коли спостерігалась лімфопенія 3 або 4 ступеня (від < 500 до 200 клітин/мм³ або < 200 клітин/мм³), порівняно з часом, коли у пацієнтів не було лімфопенії ступеня 3 або 4.
Перелік побічних реакцій
Інформація про побічні реакції, описані нижче, одержана на підставі об'єднаних даних клінічних досліджень лікування РС пероральним препаратом кладрибіну як монотерапії в кумулятивній дозі 3,5 мг/кг. База даних безпеки цих досліджень включає дані 923 пацієнтів.
Для визначення частоти побічних реакцій використовують таку термінологію: дуже поширені (≥ 1/10), поширені (від ≥ 1/100 до < 1/10), непоширені (від ≥ 1/1000 до < 1/100), поодинокі (від ≥ 1/10000 до < 1/1000), рідкісні (< 1/10000), частота невідома (не може бути встановлена на підставі наявних даних).
Інфекції та інвазії
Поширені: оральний герпес, дерматомний оперізуючий герпес.
Рідкісні: туберкульоз.
Розлади з боку крові та лімфатичної системи
Дуже поширені: лімфопенія.
Поширені: зменшення кількості нейтрофілів.
Розлади з боку шкіри та підшкірної тканини
Поширені: висипанння, алопеція.
Опис окремих побічних реакцій
Лімфопенія
У клінічних дослідженнях тимчасова лімфопенія 3 або 4 ступеня розвинулася у 20 - 25 % пацієнтів, які приймали кумулятивну дозу кладрибіну 3,5 мг/кг протягом 2 років як монотерапію. Лімфопенія 4 ступеня спостерігалася менше ніж у 1 % пацієнтів. Найбільша частка пацієнтів з лімфопенією 3 або 4 ступеня спостерігалася через 2 місяці після прийому першої дози кладрибіну кожного року (4,0 % та 11,3 % пацієнтів з лімфопенією 3 ступеня у рік 1 та рік 2, 0 % та 0,4 % пацієнтів з лімфопенією 4 ступеня у рік 1 та рік 2). Очікується, що у більшості пацієнтів кількість лімфоцитів відновиться до нормальної або лімфопенії 1 ступеня впродовж 9 місяців.
Для зменшення ризику розвитку тяжкої лімфопенії кількість лімфоцитів необхідно визначати перед, впродовж та після лікування кладрибіном, а також дотримуватися суворих критеріїв для початку та продовження лікування кладрибіном.
Злоякісні новоутворення
У клінічних дослідженнях та протягом тривалого періоду подальшого спостереження пацієнтів, які приймали кумулятивну дозу кладрибіну 3,5 мг/кг перорально, у пацієнтів, які лікувались кладрибіном, явища злоякісних новоутворень спостерігалися частіше (10 випадків на 3414 пацієнто-років [0,29 випадків на 100 пацієнто-років]) порівняно з пацієнтами, які приймали плацебо (3 випадки на 2022 пацієнто-роки [0,15 випадків на 100 пацієнто-років]).
Термін придатності. 3 роки.
Не застосовувати після закінчення терміну придатності, зазначеного на упаковці.
Умови зберігання.
Зберігати в оригінальній упаковці для захисту від вологи.
Зберігати у недоступному для дітей місці.
Упаковка. По 1, 4 або 6 таблеток в алюмінієвому блістері, запечатаному у картонну обкладинку, яку вміщують у контурну чарункову упаковку та вкладають у картонну коробку із захистом від доступу дітей.
Категорія відпуску. За рецептом.
Виробник.
НерФарМа С.Р.Л. / NerPharMa S.R.L.
Місцезнаходження виробника та адреса місця провадження його діяльності
Віале Пастер 10 (р-н Нервіано), 20014 Мілан (МІ), Італія /
Viale Pasteur 10 (loc. Nerviano), 20014 Milan (MI), Italy.
Інші медикаменти цього ж виробника
Форма: розчин для ін'єкцій по 900 МО (66 мкг)/1,5 мл по 1,5 мл у картриджі з пробкою-поршнем та рифленою кришечкою, вміщеному у ручку для введення; по 1 ручці та 20 голок у картонній коробці
Форма: порошок та розчинник для розчину для ін'єкцій, 1 або 3 флакони з порошком у комплекті з 1 або 3 флаконами з 1 мл розчинника (вода для ін'єкцій) у контурній чарунковій упаковці; по 1 контурній чарунковій упаковці в коробці; 5 флаконів з порошком у комплекті з 5 флаконами з 1 мл розчинника (вода для ін'єкцій) у контурній чарунковій упаковці; по 2 контурні чарункові упаковки в коробці
Форма: порошок для приготування розчину для ін'єкцій по 8 мг, 1 або 5 флаконів з порошком, 1 або 5 картриджів з 1,37 мл розчинника (0,3 % (м./об.) розчин м-крезолу у воді для ін’єкцій) попередньо зібраних в 1 або 5 пристроїв для розчинення (клік.ізі), що складаються з 1 або 5 корпусів пристрою та 1 або 5 стерильних перехідних канюль у картонній коробці
Форма: таблетки по 10 мг в алюмінієвому блістері № 1, № 4 або № 6 запечатаному у картонну обкладинку
Форма: розчин для ін'єкцій по 6 мг (5,83 мг/мл) по 1,03 мл у картриджах № 1