Емоклот
Реєстраційний номер: UA/17394/01/01
Імпортер: КЕДРІОН С.П.А.
Країна: ІталіяАдреса імпортера: ЛОКАЛІТА АЙ КОНТІ, КАСТЕЛЬВЕККІО ПАСКОЛІ, 55051 БАРГА, ЛУККА (ЛУ), ІТАЛIЯ
Форма
порошок та розчинник для розчину для інфузій, 500 МО/10 мл; по 500 МО у флаконі № 1 у комплекті з розчинником (вода для ін`єкцій) по 10 мл у флаконі № 1 та набором для розчинення і введення у картонній коробці
Склад
1 флакон з порошком містить фактора коагуляції крові людини VIII 500 МО
Виробники препарату «Емоклот»
Країна: Італія
Адреса: ВІА ПРОВІНСІАЛЕ (лок. БОЛОГНАНА) - 55027 ГАЛЛІКАНО (ЛУ), ІТАЛIЯ
Інструкція по застосуванню
для медичного застосування лікарського засобу
ЕМОКЛОТ
Склад
діюча речовина: фактор коагуляції крові людини VIII;
1 флакон з порошком містить фактора коагуляції крові людини VIII 500 МО або 1000 МО;
допоміжні речовини: натрію хлорид, натрію цитрат, гліцин, кальцію хлорид;
флакон з розчинником містить води для ін'єкцій 10 мл.
Відновлений розчин містить фактора коагуляції крові людини VIII 50 МО/мл (500 МО/10 мл) або 100 МО/мл (1000 МО/10 мл).
Специфічна активність препарату ЕМОКЛОТ становить приблизно 80 МО/мг білка. Активність (МО) визначається за допомогою хромогенного аналізу відповідно до Європейської фармакопеї.
Загальний вміст білка не більше 7,2 мг/флакон або 15 мг/флакон.
Препарат містить фактор фон Віллебранда людини.
Лікарська форма. Порошок та розчинник для розчину для інфузій.
Основні фізико-хімічні властивості:
порошок: білий або блідо-жовтий гігроскопічний порошок або крихка тверда речовина;
розчинник: прозора безбарвна рідина.
Фармакотерапевтична група. Антигеморагічні засоби, фактор згортання крові VIII.
Код ATХ B02B D02.
Фармакологічні властивості.
Фармакодинаміка.
Комплекс фактор VIII/ фактор фон Віллебранда складається з двох молекул (фактор VIII та фактор фон Віллебранда) з різними фізіологічними функціями.
При введенні пацієнту з гемофілією фактор VIII зв'язується з фактором фон Віллебранда, що циркулює у крові пацієнта.
Активований фактор VIII діє як кофактор для активованого фактора IX, прискорюючи перетворення фактора X в активну форму. Активований фактор X перетворює протромбін у тромбін, який, у свою чергу, перетворює фібриноген у фібрин, після чого може формуватися згусток. Гемофілія А - це пов'язане зі статтю спадкове порушення згортання крові, обумовлене зниженими рівнями фактора VIII:C, що призводить до профузних кровотеч у суглобах, м'язах або внутрішніх органах, які відбуваються спонтанно або у результаті випадкових ушкоджень чи хірургічних втручань. Замісна терапія підвищує плазмові рівні фактора VIII і, таким чином, дає змогу тимчасово відкоригувати дефіцит фактора та знизити тенденцію до кровотеч.
Фактор фон Віллебранда відіграє роль стабілізатора фактора VIII, опосередковує адгезію тромбоцитів до ділянок ушкодження судин і бере участь у агрегації тромбоцитів.
Діти
Хоча специфічні дані щодо застосування дітям відсутні, нечисленні опубліковані дані досліджень ефективності та безпеки не продемонстрували суттєвих відмінностей між дорослими та дітьми з одним і тим же захворюванням.
Фармакокінетика.
Після введення препарату приблизно 2/3-3/4 фактора VIII залишається в циркулюючій крові.
Рівень активності фактора VIII, що досягається у плазмі, становить 80-120% від розрахункової активності фактора VIII у плазмі.
Активність фактора VIII у плазмі знижується за двофазною експоненціальною кривою.
У початковій фазі розподіл між внутрішньосудинною та іншими рідинами організму відбувається з періодом напіввиведення з плазми 3-6 годин.
У наступній більш повільній фазі (яка, ймовірно, відображає поглинання фактора VIII) період напіввиведення варіює від 8 до 20 годин і у середньому становить 12 годин, що відображає істинний біологічний період напіввиведення.
Фармакокінетичні властивості препарату ЕМОКЛОТ були вивчені у клінічному дослідженні, що проводилося за участю 15 пацієнтів з тяжкою гемофілією A (з рівнем фактора VIII <1). Фармакокінетичні параметри визначалися після двох окремих інфузій (дозування 25 МО/кг), що проводилися з інтервалом 3-6 місяців. У період між двома інфузіями пацієнти отримували лікування препаратом ЕМОКЛОТ за їх звичайною терапевтичною схемою застосування (з лікувальною або профілактичною метою).
Середні величини фармакокінетичних параметрів препарату ЕМОКЛОТ, визначені під час дослідження, представлені у таблиці 1.
Таблиця 1
|
Показник |
Перша інфузія |
Друга інфузія |
||
|
Без вирахування початкових показників |
З вирахуванням початкових показників |
Без вирахування початкових показників |
З вирахуванням початкових показників |
|
|
AUC0-t (МО·мл-1·год) |
10,94 |
9,96 |
10,75 |
8,95 |
|
AUC0-¥ (МО·мл-1·год) |
13,08 |
11,22 |
12,07 |
9,89 |
|
Cltot (мл·год-1·кг-1) |
2,63 |
2,89 |
2,51 |
2,99 |
|
Поступове відновлення (%) |
2,688 |
2,671 |
||
|
t1/2a (год) |
0,543 |
0,768 |
||
|
t1/2b (год) |
12,05 |
15,16 |
Діти
Хоча специфічні дані щодо застосування дітям відсутні, нечисленні опубліковані дані досліджень фармакокінетики не продемонстрували суттєвих відмінностей між дорослими та дітьми з одним і тим же захворюванням.
Клінічні характеристики
Показання
Лікування і профілактика кровотеч у пацієнтів з гемофілією А (вроджена недостатність фактора коагуляції крові людини VIII).
Лікування набутого дефіциту фактора коагуляції крові людини VIII.
Лікування хворих на гемофілію з антитілами до фактора коагуляції крові людини VIII (інгібітори: див. також «Особливості застосування»).
Протипоказання
Підвищена чутливість до діючої речовини або до будь-якої з допоміжних речовин лікарського засобу.
Взаємодія з іншими лікарськими засобами та інші види взаємодій
Про взаємодію препаратів фактора коагуляції крові людини VIII з іншими лікарськими засобами не повідомлялося.
Особливості застосування
Підвищена чутливість
При застосуванні препарату ЕМОКЛОТ можливі алергічні реакції підвищеної чутливості.
Препарат містить сліди білків людини, відмінних від фактора VIII. У разі виникнення симптомів підвищеної чутливості пацієнтам слід рекомендувати негайно припинити застосування препарату та звернутися до лікаря. Пацієнтів слід інформувати про ранні ознаки реакцій підвищеної чутливості, включаючи висип, ґенералізовану кропив'янку, стиснення у грудній клітці, дихання з присвистом, гіпотонію та анафілаксію.
У разі виникнення шоку слід дотримуватися чинних медичних стандартів лікування шоку.
Інгібітори
Утворення нейтралізуючих антитіл (інгібіторів) до фактора коагуляції крові людини VIII є відомим ускладненням лікування пацієнтів з гемофілією А. Ці інгібітори зазвичай являють собою імуноглобуліни IgG, спрямовані проти прокоагулюючої активності фактора VIII, кількість яких визначають в одиницях Бетезда (ОБ) на 1 мл плазми за допомогою модифікованого аналізу. Ризик розвитку інгібіторів корелює зі ступенем тяжкості захворювання, а також з експозицією фактора VIII. Цей ризик найвищий впродовж перших 20 днів застосування. Рідко інгібітори можуть утворюватися після перших 100 днів застосування.
Спостерігалися випадки повторної появи інгібіторів (у низькому титрі) після переходу з одного препарату фактора VIII на інший у пацієнтів, які раніше отримували лікування з експозицією більше 100 днів з утворенням інгібіторів у анамнезі. Тому після будь-якої зміни препарату рекомендується ретельний моніторинг всіх пацієнтів щодо появи інгібіторів.
Клінічна значимість утворення інгібіторів залежить від їх титру. При низькому титрі інгібіторів, тимчасовому або постійному, ризик недостатньої клінічної відповіді нижчий, ніж при високому титрі інгібіторів.
Загалом, слід проводити ретельний моніторинг усіх пацієнтів, які отримують лікування препаратами фактора коагуляції VIII, щодо утворення інгібіторів шляхом належного клінічного спостереження та лабораторних досліджень. Якщо при застосуванні відповідної дози не досягається очікуваний рівень активності фактора VIII у плазмі або, якщо кровотеча не контролюється, слід провести дослідження на наявність інгібіторів фактора VIII. У пацієнтів з високим рівнем інгібіторів терапія фактором VIII може виявитися неефективною, і у такому випадку слід розглянути інші способи лікування. Терапію таких пацієнтів слід проводити під контролем лікаря, що має досвід лікування пацієнтів з гемофілією та інгібіторами фактора VIII.
Ускладнення, пов'язані із застосуванням катетера
У разі потреби у центральному венозному доступі слід враховувати ризик пов'язаних із застосуванням пристрою ускладнень, включаючи локальні інфекції, бактеріємію та тромбоз у місці введення катетера.
Вірусна безпека
Стандартні заходи для попередження інфекцій внаслідок застосування лікарських засобів, отриманих з крові або плазми людини, включають відбір донорів, перевірку окремих порцій і пулів плазми на специфічні маркери інфекцій та вжиття ефективних виробничих заходів для інактивації/знищення вірусів.
Незважаючи на це, при введенні лікарських засобів, виготовлених із крові або плазми людини, не можна цілком виключити можливість передачі збудників інфекції, у тому числі невідомих або нових на цей час вірусів та інших патогенів.
Заходи, яких вживають, вважаються ефективними щодо оболонкових вірусів, таких як вірус імунодефіциту людини (ВІЛ), вірус гепатиту В (HBV) і вірус гепатиту С (HCV), а також щодо необолонкових вірусів, таких як вірус гепатиту А (HAV). Заходи, яких вживають, можуть мати обмежену ефективність щодо необолонкових вірусів, таких як парвовірус В19. Інфікування парвовірусом В19 може бути серйозним для вагітних (інфікування плода) і пацієнтів з імунодефіцитом або посиленим еритропоезом (наприклад, при гемолітичній анемії).
Слід розглянути питання проведення відповідної вакцинації (від гепатиту A та B) пацієнтам, яким регулярно/повторно застосовують препарати фактора коагуляції VIII з плазми людини.
Для збереження зв'язку між пацієнтом та серією препарату настійно рекомендується щоразу при введенні препарату ЕМОКЛОТ фіксувати номер серії препарату та дані пацієнта.
Застосування у період вагітності або годування груддю.
Дослідження впливу фактора VIII на репродуктивну функцію тварин не проводилися. Частота захворювання на гемофілію А серед жінок низька, досвід застосування фактора VIII вагітним та жінкам, які годують груддю, відсутній. Тому фактор VIII слід застосовувати під час вагітності і годування груддю лише за чітких показань.
Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами.
ЕМОКЛОТ не впливає на здатність керувати транспортними засобами та працювати з іншими механізмами.
Спосіб застосування та дози
Лікування слід розпочинати під контролем лікаря, який має досвід лікування гемофілії.
Дозування
Дозування та тривалість замісної терапії залежать від ступеня тяжкості дефіциту фактора VIII, локалізації та інтенсивності кровотечі, а також від клінічного стану пацієнта.
Кількість одиниць фактора VIII, що вводиться, виражається у міжнародних одиницях (МО), що відповідає чинному стандарту ВООЗ для препаратів фактора VIII. Активність фактора VIII у плазмі виражається у відсотках (відносно нормальної плазми людини) або у міжнародних одиницях (відносно міжнародного стандарту для фактора VIII у плазмі).
Активність однієї міжнародної одиниці (МО) фактора VIII відповідає кількості фактора VIII у 1 мл нормальної плазми людини.
Лікування у разі потреби
Розрахунок необхідної дози фактора VIII ґрунтується на даних, отриманих емпіричним шляхом. 1 міжнародна одиниця (МО) фактора VIII на 1 кг маси тіла підвищує активність фактора VIII у плазмі на 1,5%-2% нормальної активності. Необхідна доза визначається за такою формулою:
Необхідна кількість одиниць =3D маса тіла (кг) ´ бажане збільшення фактора VIII (%) (МО/дл) ´ 0,4
Кількість препарату, що має бути введена, та частота введення завжди повинні бути орієнтовані на клінічну ефективність у кожному конкретному випадку.
У нижченаведених випадках кровотеч активність фактора VIII не повинна бути нижчою вказаного рівня активності у плазмі крові (в % від норми) у відповідний період. Таблицю 2 можна використовувати як інструкцію з дозування при кровотечах та хірургічних втручаннях.
Таблиця 2
Ступінь кровотечі / тип хірургічної процедури |
Необхідний рівень фактора VIII (%) (МО/дл) |
Частота введення (години) /тривалість лікування (днів) |
Кровотеча |
||
|
Ранній гемартроз, кровотеча у м'язах або кровотеча в ротовій порожнині |
20-40 |
Повторювати кожні 12-24 години щонайменше протягом 1 доби до припинення кровотечі, про що свідчить усунення болю або загоєння. |
|
Більш виражений гемартроз, кровотеча у м'язах або гематома |
30-60 |
Повторювати введення кожні 12-24 години впродовж 3-4 діб або довше, доки не будуть усунені біль та гостра недієздатність. |
|
Кровотеча, що загрожує життю |
60-100 |
Повторювати введення кожні 8-24 години до усунення загрози життю. |
Оперативні втручання |
||
|
Малі оперативні втручання, у тому числі видалення зуба |
30-60 |
Кожні 24 години принаймні 1 добу до загоєння. |
|
Великі оперативні втручання |
80-100 (до та після оперативного втручання) |
Повторювати введення кожні 8-24 години до адекватного загоєння рани; потім продовжити терапію не менше 7 діб, підтримуючи активність фактора VIII на рівні 30-60% (30-60 МО/дл). |
Профілактика
Для довготривалої профілактики кровотеч пацієнтам з тяжкою гемофілією А зазвичай призначають дози від 20 до 40 МО фактора VIII на 1 кг маси тіла з інтервалом введення від 2 до 3 діб. В окремих випадках, особливо пацієнтам молодого віку, можуть бути необхідні коротші інтервали між введеннями або вищі дози.
Протягом курсу лікування рекомендується визначати рівні фактора VIII для встановлення необхідної дози та частоти повторних введень. У разі великих оперативних втручань обов'язковим є ретельний контроль замісної терапії за допомогою аналізу коагуляції (активність фактора VIII у плазмі крові). Окремі пацієнти можуть мати різну відповідь на фактор VIII, демонструючи різні періоди напіввиведення та відновлення.
Спосіб застосування
Застосовується внутрішньовенно шляхом ін'єкції або повільної інфузії.
При внутрішньовенній ін'єкції препарат рекомендується вводити впродовж 3-5 хвилин та контролювати при цьому частоту пульсу у пацієнтів, перериваючи введення або знижуючи швидкість введення при підвищенні частоти пульсу.
Швидкість інфузії для кожного пацієнта слід визначати індивідуально.
Відновлення порошку розчинником:
1. Флакон з порошком та флакон з розчинником довести до кімнатної температури.
2. Кімнатну температуру слід підтримувати протягом всього процесу відновлення (максимум 10 хвилин).
3. Зняти захисні ковпачки з флакона з порошком та флакона з розчинником.
4. Обробити поверхні пробок на обох флаконах етиловим спиртом.
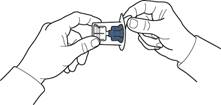
5. Відкрити упаковку пристрою, обережно відшарувавши верхню кришку, не торкаючись внутрішньої частини упаковки (рис. А).
6. Не виймати пристрій з упаковки.
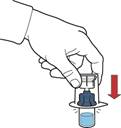
7. Перевернути упаковку з пристроєм та ввести пластиковий шип через пробку флакона з розчинником таким чином, щоб синя частина пристрою з'єдналася із флаконом з розчинником (рис. Б).
8. Утримуючи за край упаковки пристрою, видалити її, не торкаючись самого пристрою (рис. В).
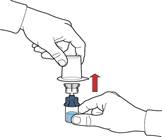
9. Переконатися, що флакон з порошком знаходиться на безпечній поверхні; перевернути систему так, щоб флакон з розчинником був над пристроєм; натиснути на прозорий адаптер на пробці флакона з порошком так, щоб ввести пластиковий шип через пробку флакона з порошком; розчинник автоматично переміститься у флакон з порошком (рис. Г).
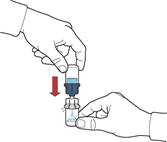
10. Після переміщення розчинника відкрутити синю частину системи, до якої під'єднаний флакон розчинника, та видалити її (рис. Д).
11. Обережно струшувати флакон до повного розчинення порошку (рис. Е).
12. Не струшувати флакон енергійно, щоб уникнути піноутворення.
Рис А |
Рис Б |
|
|
|
|
|
Рис В |
Рис Г |
|
|
|
|
|
Рис Д |
Рис Е |
|
|
|
|
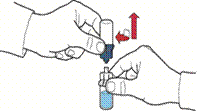
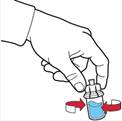
Введення розчину
Після відновлення розчин може містити незначну кількість невеликих пластівців або часток.
Перед введенням відновлений розчин слід візуально перевірити на наявність часток або зміну кольору. Розчин повинен бути прозорим або злегка опалесцентним. Не використовувати, якщо розчин каламутний або містить осад.
1. Відтягуючи поршень, заповнити шприц повітрям, під'єднати до пристрою та ввести повітря у флакон з відновленим розчином (рис. Є).
2. Зберігаючи поршень у тому ж положенні, перевернути систему догори дном так, щоб флакон з відновленим розчином знаходився над пристроєм, та повільно відтягуючи поршень, набрати концентрат у шприц (рис. Ж).
3. Від'єднати шприц, повертаючи його проти годинникової стрілки.
4. Візуально перевірити розчин у шприці, він повинен бути прозорим або злегка опалесцентним та не містити частинок.
5. Під'єднати до шприца голку-метелик та ввести препарат внутрішньовенно шляхом інфузії або повільної ін'єкції.
Рис Є |
Рис Ж |
|
|
|
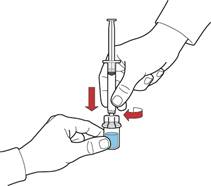
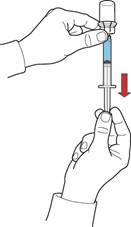
Вміст відкритих флаконів слід використати негайно.
Набраний у шприц відновлений розчин слід використати негайно.
Вміст флакона слід використовувати тільки для одного введення.
Заборонено застосування препарату після закінчення терміну придатності, вказаного на упаковці.
Будь-який невикористаний препарат або залишки слід знищити відповідно до місцевих вимог.
Діти.
Специфічні дані щодо дітей відсутні. Безпека та ефективність застосування препарату ЕМОКЛОТ дітям віком до 12 років не встановлені. Дозування підліткам (12-18 років) для кожного показання розраховується за масою тіла.
Передозування
Про симптоми передозування фактором коагуляції крові людини VIII не повідомлялося.
Побічні реакції
Підвищена чутливість або алергічні реакції (в тому числі ангіоневротичний набряк, пекучий і гострий біль у місці інфузії, озноб, припливи, ґенералізована кропив'янка, головний біль, висип, гіпотонія, летаргія, нудота, неспокій, тахікардія, стиснення у грудях, поколювання, блювання і дихання з присвистом) спостерігаються нечасто і у деяких випадках можуть прогресувати до тяжкої анафілаксії (включаючи шок).
Також спостерігалося підвищення температури.
У пацієнтів з гемофілією А, які отримують лікування препаратами фактора VIII, включаючи ЕМОКЛОТ, можуть розвинутися нейтралізуючі антитіла (інгібітори). У разі появи інгібіторів цей стан проявлятиметься як недостатня клінічна відповідь. У таких випадках рекомендується звернутися до спеціалізованого центру гемофілії.
Інформацію з безпеки щодо передачі збудників інфекцій див. у розділі «Особливості застосування».
Побічні ефекти, які можуть виникати при застосуванні фактора коагуляції крові людини VIII, представлено у таблиці 3 відповідно до класифікації систем органів MedDRA (КСО) та термінів переважного використання.
Частота оцінювалася за такими умовними категоріями: дуже часто (≥1/10); часто (≥1/100 до <1/10); нечасто (≥1/1 000 до <1/100); рідко (≥1/10 000 до <1/1 000); дуже рідко (<1/10 000), частота невідома (неможливо оцінити за наявними даними).
Підтверджені дані про частоту побічних реакцій у клінічних дослідженнях відсутні.
Нижчезазначені дані ґрунтуються на профілі безпеки фактора коагуляції крові людини VIII та частково спостерігалися у постмаркетинговому періоді (постмаркетинговий досвід застосування препарату); оскільки повідомлення про постмаркетингові побічні реакції здійснюються на добровільній основі та стосуються популяції невідомої чисельності, відсутня можливість оцінити частоту цих реакцій.
Таблиця 3
Клас системи органів MedDRA |
Побічна реакція |
Частота |
|
Розлади з боку крові та лімфатичної системи |
Поява інгібіторів фактора VIII |
Нечасто (ПРЛ) ** Дуже часто (ПРН) ** |
|
Розлади з боку імунної системи |
Підвищена чутливість |
Невідома |
|
Алергічні реакції (Підвищена чутливість)* |
Невідома |
|
|
Анафілактична реакція |
Невідома |
|
|
Анафілактичний шок |
Невідома |
|
|
Психічні розлади |
Неспокій |
Невідома |
|
Розлади з боку нервової системи |
Головний біль |
Невідома |
|
Летаргія |
Невідома |
|
|
Парестезія |
Невідома |
|
|
Розлади з боку серця |
Тахікардія |
Невідома |
|
Розлади з боку судин |
Припливи |
Невідома |
|
Гіпотонія |
Невідома |
|
|
Респіраторні, торакальні та медіастинальні розлади |
Дихання з присвистом |
Невідома |
|
Шлунково-кишкові розлади |
Нудота |
Невідома |
|
Блювання |
Невідома |
|
|
Розлади з боку шкіри та підшкірної клітковини |
Ангіоневротичний набряк |
Невідома |
|
Ґенералізована кропив'янка (кропив'янка)* |
Невідома |
|
|
Висип (кропив'янка)* |
Невідома |
|
|
Системні розлади та реакції в місці введення |
Пекучий біль у місці інфузії (біль у місці інфузії) |
Невідома |
|
Гострий біль у місці інфузії (біль у місці інфузії)* |
Невідома |
|
|
Озноб |
Невідома |
|
|
Стиснення у грудях (дискомфорт у грудях) |
Невідома |
|
|
Пірексія |
Невідома |
* Терміни MedDRA нижчого рівня, що більше відповідають опису цих побічних реакцій; у дужках наводиться термін переважного використання MedDRA.
** Дані про частоту ґрунтуються на результатах досліджень усіх препаратів фактора VIII, які включали пацієнтів з тяжкою гемофілією А. ПРЛ - пацієнти, раніше ліковані, ПРН - пацієнти, раніше не ліковані.
Діти
Специфічні дані щодо дітей відсутні.
Повідомлення про підозрювані побічні реакції
Після реєстрації лікарського засобу повідомлення про підозрювані побічні реакції є важливим, оскільки це дає змогу продовжити контролювати співвідношення користь/ризик застосування лікарського засобу. Про будь-які підозрювані побічні реакції медичним працівникам слід повідомляти через національну систему повідомлень.
Термін придатності.
3 роки.
Розчинник (вода для ін'єкцій) - 5 років.
Препарат повинен бути використаний відразу після розчинення.
Умови зберігання.
Зберігати в холодильнику (при температурі від 2 до 8 °С). Не заморожувати. Зберігати в оригінальній упаковці з метою захисту від світла, у недоступному для дітей місці.
Несумісність.
Через відсутність досліджень сумісності цей лікарський засіб не можна змішувати з іншими лікарськими засобами.
Слід використовувати лише набір для ін'єкції/інфузії, що входить до комплекту, оскільки лікування може бути неефективним через адсорбцію фактора коагуляції крові людини VIII на внутрішніх поверхнях іншого пристрою для введення.
Упаковка.
По 500 МО або 1000 МО у флаконі № 1 у комплекті з розчинником (вода для ін'єкцій) по 10 мл у флаконі №1 та набором для розчинення і введення у картонній коробці.
Категорія відпуску. За рецептом.
Виробник.
КЕДРІОН С.П.А.
Місцезнаходження виробника та його адреса місця провадження діяльності
ВІА ПРОВІНСІАЛЕ (лок. БОЛОГНАНА) - 55027 ГАЛЛІКАНО (ЛУ), Італія.
Інші медикаменти цього ж виробника
Форма: порошок та розчинник для розчину для інфузій, 1000 МО/10 мл; по 1000 МО у флаконі № 1 у комплекті з розчинником (вода для ін`єкцій) по 10 мл у флаконі № 1 та набором для розчинення і введення у картонній коробці
Форма: порошок для розчину для інфузій по 500 МО у флаконі № 1 у комплекті з розчинником (вода для ін'єкцій) по 20 мл у флаконі № 1 та набором для розчинення і введення у картонній коробці
Форма: порошок та розчинник для розчину для інфузій, 1000 МО/10 мл; по 1000 МО у флаконі № 1 у комплекті з розчинником (вода для ін`єкцій) по 10 мл у флаконі № 1 та набором для розчинення і введення у картонній коробці
Форма: порошок та розчинник для розчину для інфузій, 500 МО/10 мл; по 500 МО у флаконі № 1 у комплекті з розчинником (вода для ін`єкцій) по 10 мл у флаконі № 1 та набором для розчинення і введення у картонній коробці
Форма: порошок та розчинник для розчину для інфузій, 500 МО/10 мл; по 500 МО у флаконі № 1 у комплекті з розчинником (вода для ін`єкцій) по 10 мл у флаконі № 1 та набором для розчинення і введення у картонній коробці