Цитафін
Реєстраційний номер: UA/16204/01/01
Імпортер: Емкур Фармасьютікалс Лтд
Країна: ІндіяАдреса імпортера: Т-184, M.I.Д.C., Бхосарі, Пуне-411026, Індія
Форма
ліофілізат для розчину для інфузій по 1000 мг у флаконах № 1
Склад
1 флакон містить гемцитабіну гідрохлориду еквівалентно гемцитабіну 1000 мг
Виробники препарату «Цитафін»
Країна: Індія
Адреса: Плот № П-2, ІТ Парк, Фаза ІІ MIДC, Хіндіваді, Пуне, ІН 411 057, Індія
Інструкція по застосуванню
для медичного застосування лікарського засобу
ЦИТАФІН
(СITAFINE)
Склад
діюча речовина: гемцитабін;
1 флакон містить гемцитабіну гідрохлориду еквівалентно гемцитабіну 1000 мг;
допоміжні речовини: маніт (Е 421), натрію ацетат безводний.
Лікарська форма. Ліофілізат для розчину для інфузій.
Основні фізико-хімічні властивості: ліофілізована маса або порошок білого або майже білого кольору.
Фармакотерапевтична група.
Протипухлинні лікарські засоби. Структурні аналоги піримідину.
Код АТХ L01B С05.
Фармакологічні властивості
Фармакодинаміка.
Цитотоксична активність у клітинних культурах.
Гемцитабін проявляє значну цитотоксичну дію на різні види людських клітин раку та культивовані мишачі клітини раку. Гемцитабін має клітинно-фазову специфічність, головним чином знищуючи клітини, що проходять фазу синтезу ДНК (S-фаза), а за певних умов блокує проходження клітин через межу фази G1/S. Цитотоксична дія гемцитабіну in vitro залежить від концентрації та часу.
Протипухлинна дія на доклінічних моделях.
На тваринних моделях пухлин протипухлинна дія гемцитабіну залежить від графіка введення. При щоденному введенні гемцитабіну спостерігалась висока летальність серед тварин та мінімальна протипухлинна дія. Проте при введенні гемцитабіну на кожен 3-й чи 4-й день у нелетальних дозах він проявляє значну протипухлинну дію проти широкого спектра пухлин у мишей.
Механізм дії.
Внутрішньоклітинний метаболізм та механізм дії.
Гемцитабін (dFdC) - піримідиновий антиметаболіт, який метаболізується внутрішньоклітинно під впливом нуклеозидкінази до активних дифосфатних (dFdCDP) і трифосфатних (dFdCTP) нуклеозидів. Виявляється, що цитотоксична дія гемцитабіну зумовлена інгібуванням синтезу ДНК двома активними метаболітами - дифосфатним і трифосфатним нуклеозидами. По-перше, дифосфатний нуклеозид інгібує рибонуклеотидредуктазу, що каталізує реакції, внаслідок яких утворюються дезоксинуклеозидтрифосфати (dCTP) для синтезу ДНК. Інгібування цього ферменту спричиняє зменшення концентрації дезоксинуклеозидів узагалі і, зокрема, концентрації дезоксинуклеозидтрифосфатів. По-друге, dFdCTP конкурують з dCTP при побудові ДНК (самопотенціювання).
Крім того, невелика кількість гемцитабіну може приєднуватись до РНК. Таким чином, зменшення внутрішньоклітинної концентрації dCTP посилює приєднання трифосфатних нуклеозидів до ланцюга ДНК. Іпсилон ДНК-полімерази неспроможні усувати гемцитабін і відновлювати ланцюги ДНК, що синтезуються. Після приєднання внутрішньоклітинних метаболітів гемцитабіну до ланцюгiв ДНК, які синтезуються, долучається один додатковий нуклеотид, що призводить до повного інгібування подальшого синтезу ДНК (приховане закінчення ланцюга) і зaпрограмованої загибелі клітини, відомої як апоптоз.
Фармакокінетика.
Серед пацієнтів, що брали у часть у фармакокінетичних дослідженнях приблизно у 45 % було діагностовано недрібноклітинний рак легенів, а у 35 % пацієнтів - рак підшлункової залози. Наступні фармакокінетичні параметри було отримано при застосуванні доз у діапазоні від 500 до 2592 мг/м2, які вводилися шляхом інфузії впродовж часового інтервалу від 0,4 до 1,2 години.
Пікові концентрації у плазмі крові (дані отримані за 5 хвилин до завершення введення інфузії) становили від 3,2 до 45,5 мкг/мл. Концентрації у плазмі крові основного компонента після введення дози концентрацією 1000мг/м2 впродовж 30 хвилин перевищували 5мгк/мл через приблизно 30 хвилин після завершення інфузії та перевищували 0,4 мкг/мл ще через 1 годину.
Розподіл.
Об'єм розподілу в центральній камері становить 12,4 л/м2 у жінок і 17,5 л/м2 - у чоловіків (міжіндивідуальна варіабельність становить 91,9 %). Об'єм розподілу в периферичній камері становив 47,4 л/м2 і не залежав від статі. Зв'язування з протеїнами плазми крові незначне і ним можна знехтувати.
Період напіввиведення.
Залежно від віку та статі період напіввиведення становить від 42 до 94 хв. При застосуванні препарату в рекомендованих дозах процес виведення гемцитабіну майже повністю завершується через 5-11 годин від початку введення інфузії. При застосуванні гемцитабіну 1 раз на тиждень препарат не акумулюється.
Метаболізм.
Гемцитабін швидко метаболізується цитидиндезаміназою у печінці, нирках, крові та в інших тканинах. Внаслідок внутрішньоклітинного метаболізму гемцитабіну утворюються гемцитабіну моно-, ди- і трифосфати (dFdCMP, dFdCDP і dFdCTP відповідно), при цьому активними метаболітами вважаються dFdCDP і dFdCTP. Ці внутрішньоклітинні метаболіти не виявляються у плазмі крові або сечі. Основний метаболіт 2'-дезокси-2',2'-дифторуридин (dFdU) - неактивний і виявляється у плазмі крові і сечі.
Виведення.
Системний кліренс варіюється у діапазоні від 29,2 л/год/м2 до 92,2 л/год/м2, залежно від віку і статі (міжіндивідуальна варіабельність становить 52,2 %). Кліренс у жінок приблизно на 25 % нижчий за такий показник у чоловіків. Незважаючи на швидкість кліренсу, він знижується з віком у чоловіків і жінок. При застосуванні гемцитабіну в рекомендованій дозі 1000 мг/м2 у вигляді інфузії впродовж 30 хв. нижчий кліренс у жінок і чоловіків не є приводом для зменшення дози гемцитабіну.
Виведення із сечею. Менше 10 % препарату виділяється в незмінному вигляді.
Нирковий кліренс становив 2-7 л/ч/м2.
Впродовж тижня після введення препарату виводиться від 92 % до 98 % введеної дози, 99 % виводиться із сечею, в основному у вигляді dFdU і 1% від введеної дози виводиться з калом.
Комбінована терапія гемцитабіну з паклітакселом.
Комбінована терапія гемцитабіну з паклітакселом не впливає на фармакокінетику жодного з цих лікарських засобів.
Комбінована терапія гемцитабіну з карбоплатином.
Комбінована терапія гемцитабіну з карбоплатином не впливає на фармакокінетику гемцитабіну.
Порушення функцій нирок.
Ниркова недостатність помірного або середнього ступеня (швидкість клубочкової фільтрації від 30 мл/хв до 80 мл/хв) не має тривалого значного впливу на фармакокінетику гемцитабіну.
Клінічні характеристики
Показання
Рак сечового міхура. Гемцитабін у комбінації з цисплатином показаний для лікування хворих на локально рецидивуючий чи метастатичний рак сечового міхура.
Рак підшлункової залози. Гемцитабін показаний для лікування пацієнтів із локально прогресуючими чи метастатичними аденокарциномами підшлункової залози.
Рак легeнiв недрібноклітинний. Гемцитабін у комбінації з цисплатином показаний як препарат першої лінії для лікування пацієнтів з локально прогресуючим чи метастатичним недрібноклітинним раком легень. Гемцитабін як монотерапія показаний для лікування пацієнтів літнього віку та пацієнтів із другим функціональним статусом.
Рак яєчників. Гемцитабін у комбінації з карбоплатином показаний для лікування пацієнтів з локально прогресуючою чи метастатичною епітеліальною карциномою яєчників. Препарат показаний для лікування пацієнтів із рецидивом епітеліальної карциноми яєчників після періоду ремісії, що становив не менше 6 місяців, після попередньої терапії у першій лінії препаратами платини.
Рак молочної залози. Гемцитабін у комбінації з паклітакселом показаний для лікування хворих на неоперабельний, локально рецидивуючий чи метастатичний рак молочної залози після попередньої ад'ювантної/неоад'ювантної хіміотерапії. Попередня хіміотерапія повинна включати антрациклін, якщо немає протипоказань.
Рак жовчних протоків. Гемцитабін показаний для лікування хворих на рак жовчних протоків.
Протипоказання
Підвищена чутливість до діючої речовини або до будь-якої з допоміжних речовин препарату.
Період годування груддю.
Особливі заходи безпеки
Особливості пpuготування розчину для інфузії.
Як і у випадку з іншими цитостатиками, слід приділяти велику увагу приготуванню та застосуванню розчину для інфузій. Приготування розчину для інфузій потрібно проводити у захисному боксі та з використанням рукавичок і захисних плащів. Якщо робота у захисному боксі неможлива, необхідно використовувати маску та захисні окуляри.
Потрапляння розчину в очі може спричинити сильне подразнення. У такому випадку необхідно негайно ретельно промити очі водою. Якщо подразнення не зникає, потрібно звернутись до лікаря. У випадку потрапляння розчину на шкіру негайно промити шкіру водою.
Взаємодія з іншими лікарськими засобами та інші види взаємодій
Специфічних досліджень взаємодії не проводилося.
Радіотерапія.
Супутня радіотерanія (разом або ≤ 7 днів після). Токсичність, спричинена терапією різними методами, залежить від багатьох факторів, включаючи дозу гемцитабіну, частоту інфузій, дозу радіації, використовувану техніку, зону та обсяг опромінювання.
Доклінічні та клінічні дослідження показали, що гемцитабін має радіосенсибілізуючу активність. У ході одного випробування, де гемцитабін у дозі 1000 мг/м2 вводили протягом періоду до 6 тижнів разом із терапевтичним опромінюванням грудної клітки пацієнтам з недрібноклiтинним раком легенів, спостерігалася значна токсичність у вигляді тяжкого і потенційно загpозливого для життя пацієнта мукозиту, зокрема у вигляді езофагітy та пневмоніту, особливо у пацієнтів, для лікування яких застосовували радіотерапію у великих дозах (медіана лікування обсягом 4,795 см3). У ході наступних досліджень було запропоновано, що доцільно вводити гемцитабін у нижчих дозах із супутньою радіотерапією із очікуваною токсичністю, як було зроблено в ході дослідження II фази недрібноклітинного раку легень, де опромінювання грудної клітки в дозі 66 Gy застосовувалося разом із введенням гемцитабіну (600 мг/м2, чотири рази) та цисплатину (80 мг/м2, двічі) впродовж 6 тижнів. Оптимальний режим безпечного застосування гемцитабіну з терапевтичними дозами опромінення ще не визначений для всіх типів пухлин.
Несупутня радіотерапія (> 7 днів). Аналіз даних не виявив підвищення токсичності при застосуванні гемцитабіну понад 7 днів до чи після опромінення, крім випадків прояву «радіаційної пам'яті». Дані показують, що застосування гемцитабіну можна починати після того, як гострі ефекти опромінення минають або щонайменше через тиждень після радіотерапії.
Повідомлялося про ушкодження тканин опромінених зон після радіотерапії (наприклад езофагіти, коліти та пневмоніти) при застосуванні як із супутнім, так і з несупутнім призначенням гемцитабіну.
Інші.
Сумісне застосування живих ослаблених вакцин, в тому числі вакцини проти жовтої лихоманки, не рекомендується через ризик виникнення системного, можливо, летального захворювання, зокрема у пацієнтів із імуносупресією.
Особливості застосування
Збільшення тривалості інфузії і частоти введення доз підвищують токсичність.
Гематологічна токсичність.
Гемцитабін може послаблювати функцію кісткового мозку, що проявляється лейкоцитопенією, тромбоцитопенією та анемією.
Пацієнтам, які одержують гемцитабін, перед кожною дозою необхідно перевіряти кількість тромбоцитів, лейкоцитів і гpанулоцитiв. Доза препарату може зменшуватись або можна відкласти введення дози у разі виявлення пригнічення кісткового мозку (мієлосупресії). У той же час мієлосупресія є короткотривалою та найчастіше не призводить до зменшення дози або припинення терапії.
Кількість периферичних клітин крові може знижуватись і після припинення терапії гемцитабіном. У пацієнтів із порушеною функцією кісткового мозку необхідно з обережністю призначати лікування. Як і при лікуванні іншими цитотоксичними агентами, необхідно зважати на ризик виникнення кумулятивного пригнічення кісткового мозку у випадку призначення гемцитабіну з іншими препаратами для хіміотерапії.
Печінкова недостатність. Препарат із обережністю призначаюти пацієнтам з печінковою і нирковою недостатністю, оскільки в ході клінічних досліджень одержано недостатньо даних, щоб рекомендувати точні дози для таких пацієнтів. Введення гемцитабіну при метастазах у печінці, при гепатиті та алкоголізмі в анамнезі, а також при цирозі печінки може призвести до збільшення печінкової недостатності. Періодично слід проводити лабораторну оцінку ниркових та печінкових показників (включаючи вірусологічні дослідження).
Супутня радіотерaпія.
В ході супутньої радіотерапії (разом або ≤ 7 днів після) повідомлялося про токсичність.
Живі вакцини.
Не рекомендується застосування вакцини проти жовтої гарячки та інших живих ослаблених вакцин пацієнтам, які отримують лікування гемцитабіном.
Синдром зворотної задньої енцефалопатії.
Повідомлялося про випадки розвитку синдрому зворотної задньої енцефалопатії (PRES) із потенційно тяжкими наслідками у пацієнтів, які отримували лікування гемцитабіном в якості монотерапії або в комбінації з іншими хіміотерапевтичними препаратами. У більшості пацієнтів, які отримували гемцитабін та у яких повідомлялося про випадки синдрому зворотної задньої енцефалопатії (PRES), спостерігалися гостра гіпертензія та епілептичні напади, також у пацієнтів могли бути і інші симптоми, такі як головний біль, летаргія, сплутаність свідомості та втрата зору.
Вищевказаний стан (синдром) діагностують за допомогою магнітно-резонансної терапії (МРТ). Синдром зворотної задньої енцефалопатії (PRES) є зворотнім станом за умови застосування належних заходів підтримуючої терапії. Якщо синдром зворотної задньої енцефалопатії (PRES) розвивається в ході терапії гемцитабіном, слід припинити терапію та розпочати проведення підтримуючих заходів, зокрема здійснення контролю артеріального тиску, протисудомна терапія.
Серцево-судинна система.
Через ризик розвитку серцевих або судинних порушень, пов'язаних із застосуванням гемцитабіну, особливу увагу слід приділяти при призначенні препарату пацієнтами із серцево-судинними захворюваннями в анамнезі.
Cиндром «капілярного просочування».
Повідомлялося про синдром «капілярного просочування» у пацієнтів, які отримували гемцитабін при монотерапії або при комбінованому застосуванні з іншими препаратами для хіміотерапії. При умові завчасного виявлення та застосування відповідної терапії синдром «капілярного просочування» зазвичай піддається лікуванню, але повідомлялось і про летальні наслідки. Цей стан виникає через підвищену системну судинну проникність, при якій рідина та протеїни з внутрішньосудинного простору просочуються в інтерстицій. Повідомлялося про наступні клінічні ознаки: генералізований набряк, збільшення ваги, гіпоальбумінемія, тяжка форма гіпотензії, гостра ниркова недостатність, набряк легенів. Введення препарату потрібно припинити при появі перших ознак синдрому «капілярного просочування» та застосувати відповідну терапію. Синдром «капілярного просочування» може з'явитися на пізніх циклах, його зазвичай пов'язують з дистрес-синдромом у дорослих.
Респіраторна система.
Повідомлялось про вплив на легені, інколи дуже сильний (такий як набряк легенів, інтерстиціальний пневмоніт або респіраторний дистрес синдром дорослих (РДСД)). Якщо такі явища розвиваються, слід подумати про припинення лікування препаратом. Поліпшити стан можна, завчасно вживши заходів симптоматичної терапії.
Видільна та сечостатева система.
Гемолітико-уремічний синдром (ГУС.)
Клінічні дані, пов'язані з гемолітико-уремічним синдромом (ГУС), рідко відзначались у післяреєстраційних дослідженнях у пацієнтів, які отримували гемцитабін. Гемолітико-уремічний синдром (ГУС) потенційно небезпечний для життя. Введення препарату потрібно припинити при появі перших ознак будь-якого доказу мікроангіопатичної гемолітичної aнeмiї, наприклад, при швидкому зниженні вмістy гемоглобіну з супровідною тромбоцитопенією, підвищенні рівня білірубіну сироватки крові, креатиніну сироватки крові, сечовини крові чи лактатдегідрогенази. Ниркова недостатність може не бyти оборотною навіть у разі припинення терапії і може з'явитися потреба в діалізі.
Фертильність.
У ході досліджень фертильності гемцитабін спричиняв у мишей-самців гіпосперматогенез. Таким чином, чоловікам, які отримують лікування гемцитабіном, не рекомендується планувати народження дітей в ході та впродовж 6 місяців після терапії. Зважаючи на можливість втрати фертильності внаслідок терапії гемцитабіном, чоловікам рекомендується вжити заходів щодо зберігання сперми перед початком лікування.
Натрій.
1 флакон препарату містить 17,5 мг (<1 ммоль) натрію, тобто натрій майже відсутній.
Застосування у період вагітності або годування груддю.
Вагітність.
Немає належних даних щодо застосування гемцитабіну вагітним. Дослідження на тваринах показали репродуктивну токсичність. Враховуючи результати досліджень на тваринах та механізм дії, не слід застосовувати гемцитабін у період вагітності, крім випадків очевидної необхідності. Необхідно рекомендувати жінкам не вагітніти в ході лікування гемцитабіном та повідомляти лікаря про те, що вони завагітніли під час застосування гемцитабіну.
Годування груддю.
Невідомо, чи проникає гемцитабін у грудне молоко і не виключено появу побічних реакцій у немовлят, що знаходяться на грудному вигодовуванні. Тому слід припинити годування груддю впродовж лікування гемцитабіном.
Фертильність.
У ході досліджень фертильності гемцитабін спричиняв у мишей-самців гіпосперматогенез. Таким чином, чоловікам, які отримують лікування гемцитабіном, не рекомендується планувати народження дітей в ході та впродовж 6 місяців після терапії. Зважаючи на можливість втрати фертильності внаслідок терапії гемцитабіном, чоловікам рекомендується вжити заходів щодо зберігання сперми перед початком лікування.
Здатність впливати на швидкість реакції при керуванні автотранспортом або роботі з іншими механізмами.
Жодних досліджень щодо вивчення здатності впливати на швидкість реакції при керуванні автотранспортом або роботи з іншими механізмами не проводилось. Оскільки гемцитабін може спричиняти сонливість, від легкої до помірної, особливо у комбінації з алкоголем, пацієнтам необхідно уникати експлуатації технічних засобів, керування автомобілем, поки вищезазначене явище не зникне.
Спосіб застосування та дози
Гемцитабін застосовує лише лікар, який має досвід протиракової хіміотерапії.
Рекомендовані дози.
Рак сечового міхура.
Комбіноване застосування. Дорослі. Рекомендована доза Цитафіну - 1000 мг/м2, що вводиться шляхом внутрішньовенної 30-хвилинної інфузії. Цю дозу слід давати у 1-й, 8-й і 15-й дні кожного 28-денного циклу у комбінації з цисплатином. Цисплатин дається рекомендованою дозою 70 мг/м2 у 1-й день після Цитафіну або в 2-й день кожного 28-денногo циклу. Потім цей 4-тижневий цикл повторюється. Зменшення дози з кожним циклом або впродовж якогось 1 циклу можна застосовувати залежно від ступеня тoксичності, якої зазнає пацієнт.
Рак підшлункової залози.
Дорослі. Рекомендована доза Цитафіну становить 1000 мг/м2, що вводиться шляхом внyтpішньовенногo вливання впродовж 30 хв. 1 раз на тиждень впродовж 7 тижнів, після чого робиться тижнева перерва. Наступні цикли складаються зі щотижневиx інфузій впродовж 3 тижнів поспіль з перервою кожного 4-го тижня. 3меншення дози з кожним циклом або впродовж якогось 1 циклу може відбуватися залежно від ступеня токсичності, якої зазнає пацієнт.
Рак легенів недрібноклітинний.
Монотерапія. Дорослі. Рекомендована доза становить 1000 мг/м2 і вводиться шляхом 30-хвилинної внутрішньовенної інфузії 1 раз на тиждень впродовж 3 тижнів, після чого робиться однотижнева перерва. Чотиритижневий цикл повторюється. Зменшення дози з кожним циклом або впродовж якогoсь 1 циклу можна проводити залежно від ступеня токсичності, якої зазнає пацієнт.
Комбіноване застосування. Дорослі. Рекомендована доза становить 1250 мг/м2 поверхні тіла та вводиться шляхом внутрішньовенної інфузії впродовж 30 хв. у 1-й та 8-й дні кожного 21-денного циклу. Доза препарату може зменшуватися з кожним циклом або впродовж якогось 1 циклу залежно від ступеня токсичності, якої зазнає хворий. Цисплатин вводити рекомендованою дозою 75-100 мг/ м2 1 раз на 3 тижні циклу.
Рак молочної залози.
Комбіноване застосування.
Дорослі. Цитафін у комбінації з
паклітакселом рекомендовано вводити у такому режимі:
паклітаксел (175 мг/м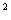 ) вводити у 1-й день впродовж
3-годинної внутрішньовенної інфузії, після нього вводити
гемцитабін (1250 мг/м) впродовж 30-хвилинної
внутрішньовенної інфузії у 1-й і 8-й дні кожного 21-денного
циклу. Доза препарату може зменшуватися з кожним циклом
або впродовж якогось
1 циклу залежно від ступеня токсичності,
якої зазнає хворий. Перед першим введенням комбінації
гемцитабіну та паклітакселу у пацієнтів має бути абсолютна
кількість гранулоцитів щонайменше 1,500
(х10
) вводити у 1-й день впродовж
3-годинної внутрішньовенної інфузії, після нього вводити
гемцитабін (1250 мг/м) впродовж 30-хвилинної
внутрішньовенної інфузії у 1-й і 8-й дні кожного 21-денного
циклу. Доза препарату може зменшуватися з кожним циклом
або впродовж якогось
1 циклу залежно від ступеня токсичності,
якої зазнає хворий. Перед першим введенням комбінації
гемцитабіну та паклітакселу у пацієнтів має бути абсолютна
кількість гранулоцитів щонайменше 1,500
(х10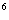 /л).
/л).
Рак яєчників.
Комбіноване застосування.
Дорослі. Цитафін у комбінації з карбоплатином рекомендовано
вводити у дозах: гемцитабін 1000
мг/м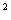 шляхом 30-хвилинного
внутрішньовенного вливання у 1-й
та 8-й дні 21-денного
циклу. У 1-й день циклу після Цитафіну вводити карбоплатин
у дозі, що забезпечує AUC 4 мг/мл*хв. Дозу препарату можна
зменшувати з кожним циклом або впродовж
якогось одного циклу залежно від ступеня токсичності,
якої зазнає хворий.
шляхом 30-хвилинного
внутрішньовенного вливання у 1-й
та 8-й дні 21-денного
циклу. У 1-й день циклу після Цитафіну вводити карбоплатин
у дозі, що забезпечує AUC 4 мг/мл*хв. Дозу препарату можна
зменшувати з кожним циклом або впродовж
якогось одного циклу залежно від ступеня токсичності,
якої зазнає хворий.
Рак жовчних протоків.
Монотерапія. Дорослі. Рекомендована доза Цитафіну - 1000 мг/м2, що слід вводити внутрішньовенно впродовж 30 хв. Інфузію проводити 1 раз на тиждень 3 тижні поспіль, потім 1 тиждень перерва. Цей чотиритижневий цикл повторювати. Зменшення дози з кожним циклом або впродовж якогось 1 циклу може відбуватися залежно від величини токсичності, якої зазнає пацієнт.
Комбіноване застосування. Дорослі. Цитафін у комбінації з цисплатином: рекомендується застосовувати цисплатин 70 мг/м2 у 1-й день циклу шляхом внутрішньовенної інфузії, далі вводити Цитафін у дозі 1250 мг/м2. Цитафін вводити в 1-й та 8-й дні кожного 21-денного циклу шляхом 30-хвилинної внутрішньовенної інфузії. Цей 3-тижневий цикл повторювати. Зменшення дози з кожним циклом або впродовж якогось 1 циклу можна застосовувати залежно від ступеня тoксичності, якої зазнає пацієнт.
Контроль токсичності та модифікація дози, пов'язана з токсичністю.
Модифікація дози, пов'язана з негематологічною токсичністю.
Для виявлення негематологічної токсичності необхідно здійснювати періодичне об'єктивне обстеження та перевірку функцій нирок і печінки. Зменшення дози з кожним циклом або впродовж якогось 1 циклy можна проводити залежно від ступеня токсичності, якої зазнає пацієнт.
Загалом, при виявленні негематологічної токсичності значного ступеня (Ступінь ІІІ або IV), крім нудоти або блювання, дозу Цитафіну можна зменшувати або можна відкласти введення дози при наявності гематологічної токсичності на розсуд лікаря. Поки, на думку лікаря, токсичність не буде скоригована, від лікування слід утриматися.
Модифікація дози, пов'язана із гематологічною токсичністю.
На початку циклу лікування.
У пацієнтів, які застосовують Цитафін, перед кожною дозою слід перевіряти кількість тромбоцитів і гранулоцитів. Абсолютна кількість гранулоцитів перед початком циклу повинна становити не менше 1500 (х106/л), а тромбоцитів - 100000 (х106/л).
Впродовж циклу лікування.
У разі необхідності дозу Цитафіну можна зменшувати або можна відкласти введення дози при наявності гематологічної токсичності відповідно до такої градації:
|
Модифікація дози Цитафіну впродовж циклу лікування за показаннями: рак сечового міхура, недрібноклітинний рак легенів, рак підшлункової залози при монотерапії або при комбінованому застосуванні з цисплатином |
|||
|
Абсолютна кількість гранулоцитів (х106/л) |
Кількість тромбоцитів (х106/л) |
Відсоток стандартної дози Цитафіну (%) |
|
|
> 1000 500-1000 < 500 |
та чи чи |
> 100000 50000-100000 < 50000 |
100 75 відкласти введення дози* |
*Від введення дози впродовж циклу слід утриматися, поки абсолютна кількість гранулоцитів не досягне значення не менше 500(х106/л), а тромбоцитів - 50000 (х106/л).
|
Модифікація дози Цитафіну впродовж циклу лікування за показаннями: рак молочної залози при комбінованому застосуванні з паклітакселем |
|||
|
Абсолютна кількість гранулоцитів (х106/л) |
Кількість тромбоцитів (х106/л) |
Відсоток стандартної дози Цитафіну (%) |
|
|
> 1200 1000-1200 700-1000 < 700 |
та чи та чи |
> 75000 50000-75000 ≥ 50000 < 50000 |
100 75 50 відкласти введення дози* |
*Введення дози не буде відновлено впродовж циклу. Лікування буде розпочато з першого дня наступного циклу, як тільки абсолютна кількість гранулоцитів досягне значення не менше 1500 (х106/л), а тромбоцитів - 100000 (х106/л).
|
Модифікація дози Цитафіну впродовж циклу лікування за показаннями: рак яєчників при комбінованому застосуванні з карбоплатином |
|||
|
Абсолютна кількість гранулоцитів (х106/л) |
Кількість тромбоцитів (х106/л) |
Відсоток стандартної дози Цитафіну (%) |
|
|
> 1500 1000- 1500 < 1000 |
та чи чи |
≥ 100000 75000-100000 < 75000 |
100 50 відкласти введення дози* |
*Введення дози не буде відновлено протягом циклу. Лікування буде розпочато з першого дня наступного циклу, як тільки абсолютна кількість гранулоцитів досягне значення не менше 1500 (х106/л), а тромбоцитів - 100000 (х106/л).
Модифікація дози, пов'язана з гематологічною токсичністю впродовж наступних циклів, для усіх показань.
Дозу Цитафіну необхідно знизити до 75 % від початкової дози, що вводилась на початку лікування, у випадку наступних проявів гематологічної токсичності:
- Абсолютна кількість гранулоцитів < 500 х 106/л впродовж понад 5 діб.
- Абсолютна кількість гранулоцитів < 100 х 106/л впродовж понад 3 діб.
- Фебрильна нейтропенія.
- Кількість тромбоцитів < 25 000 х 106/л.
- Відкладення циклу у зв'язку із проявами токсичності більше ніж на 1 тиждень.
Метод застосування.
Цитафін добре переноситься впродовж проведення інфузії і його можна вводити при амбулаторному лікуванні. У випадку виникнення гематоми необхідно негайно зупинити введення інфузії та продовжити введення в іншу судину. Необхідно ретельно контролювати стан пацієнта після проведення інфузії.
Особливі групи пацієнтів.
Пацієнти з печінковою і нирковою недостатністю. Препарат з обережністю призначаюти пацієнтам із печінковою і нирковою недостатністю, оскільки в ході клінічних досліджень одержано недостатньо даних, щоб рекомендувати точні дози для таких пацієнтів.
Пацієнти літнього віку (˃ 65 років). Препарат добре переноситься пацієнтами віком від 65 років. Немає підстав вважати, що необхідні коригування дози пацієнтам літнього віку, крім тих, які вже рекомендовані для всіх пацієнтів.
Інструкції з приготування розчину (та подальшого розведення, якщо це необхідно). Єдиним випробуваним розчинником для розчинення препарату є 0,9 % розчин натрію хлориду для ін'єкцій без консервантів.
Відповідно до значень розчинності максимальна концентрація для Цитафіну після приготування розчину становить 40 мг/мл. Відновлення у концентраціях, що перевищують 40 мг/мл, може призвести до неповного розчинення препарату, і цього слід уникати.
1. Приготування розчину та подальше його розведення потрібно здійснювати в асептичних умовах.
2. Для приготування розчину додати не мeнше 25 мл 0,9 % розчину натрію хлориду для ін'єкцій у флакон, що містить 1000 мг порошку гемцитабіну. Це забезпечує концентрацію гемцитабіну 38 мг/мл, що також враховує об'єм заміщення ліофілізату. Збовтати, щоб розчинити. Можливе подальше розведення приготованого розчину 0,9 % розчином натрію хлориду для ін'єкцій без консервантів. Відповідну кількість лікарського препарату можна вводити одразу після пригoтyвання або ще розвести 0,9 % розчином натрію хлориду для ін'єкцій. Отриманий розчин може бути прозорим або злегка жовтуватим.
3. Засоби для парентерального введення необхідно оглядати перед введенням візуально на наявність сторонніх часток і зміну забарвлення. У разі наявності сторонніх часток розчин не можна застосовувати. Будь-які невикористані частини лікарського засобу або відходи слід знищити відповідно до чинного законодавства.
Діти.
Гемцитабін не рекомендовано застосовувати дітям у зв'язку з тим, що недостатньо даних з ефективності та безпеки у цій групі пацієнтів.
Передозування
Відомого антидоту на випадок передозування гемцитабіну немає.
Клінічно допустима токсичність спостерігалася при призначенні дози до 5700 мг/м2 шляхом 30-хвилинної внутрішньовенної інфузії кожні 2 тижні.
У випадку підозри на передозування необхідно здійснювати контроль стану пацієнта, проводити відповідні аналізи крові, у разі необхідності призначати симптоматичну терапію.
Побічні реакції
Побічні реакції, пов'язані з лікуванням гемцитабіном, про які найчастіше повідомлялося: нудота, як з блюванням так і без, підвищення рівня печінкових трансаміназ (АлАт та АсАт), а також лужної фосфатази спостерігалися у приблизно 60 % пацієнтів; про протеїнурію та гематурію повідомлялося приблизно у 50 % пацієнтів; задишка спостерігалася у 10-40 % пацієнтів (найбільша частота спостерігалась у хворих на рак легень); алергічні висипання на шкірі спостерігалися у 25 % пацієнтів, а у 10 % вони супроводжувалися свербежем.
Частота появи та сила побічних реакцій залежать від дози, швидкості введення, інтервалів між дозами. Дозозалежні побічні реакції включають зниження рівня тромбоцитів, лейкоцитів та гранулоцитів.
Дані, отримані в ході клінічних досліджень.
Наступна таблиця з побічними реакціями та частотою появи отримана в ході клінічних досліджень. В кожній групі побічні реакції надані в порядку зменшення серйозності, з частотою: дуже часто (≥ 1/10), часто (≥ 1/100 і < 1/10), нечасто (≥ 1/1000 і < 1/100), рідко (≥ 1/10000 і < 1/1000), дуже рідко (<1/10000).
|
Органи та системи |
Частота |
|
З боку кровотворної та лімфатичної системи |
Дуже часто · Лейкопенія (Нейтропенія III ступеня =3D 19,3%; IV ступеня =3D 6% ) Пригнічення функції кісткового мозку найчастіше є за силою від незначного до помірного та найбільше впливає на кількість гранулоцитів · Тромбоцитопенія · Анемія Часто · Фебрильна нейтропенія Дуже рідко · Тромбоцитоз |
|
З боку імунної системи |
Дуже рідко · Анафілактоїдна реакція |
|
Розлади метаболізму та харчування |
Часто · Анорексія |
|
З боку нервової системи |
Часто · Головний біль · Сонливість · Безсоння Нечасто · Порушення мозкового кровообігу Дуже рідко
|
|
З боку серцево-судинної системи |
Нечасто · Аритмії, найчастіше суправентрикулярні за походженням · Серцева недостатність Рідко · Інфаркт міокарда · Клінічні прояви периферичного васкуліту та гангрени · Артеріальна гіпотензія Дуже рідко · Синдром «капілярного просочування» |
|
З боку респіраторної системи, органів грудної
клітки та середостіння
|
Дуже часто · Задишка (найчастіше легка та проходить без лікування) Часто · Кашель · Риніт Нечасто · Інтерстиціальний пневмоніт · Бронхоспазм (найчастіше легкий та транзиторний, але може виникнути необхідність парентерального лікування) Рідко · Набряк легень · Дистрес-синдром у дорослих |
|
З боку травної системи |
Дуже часто · Нудота · Блювання Часто · Діарея · Стоматит та поява виразок у ротовій порожнині · Запор Дуже рідко · Ішемічний коліт |
|
З боку гепатобіліарної системи |
Дуже часто · Підвищення рівнів печінкових ферментів, таких як аспартат-амінотрансфераза (АSТ), аланін-амінотрансфераза (АLТ) та лужна фосфатаза Часто · Підвищення рівнів білірубіну Нечасто · Серйозна гепатотоксичність, що призводить до печінкової недостатності та летального наслідку Рідко · Підвищення рівнів гамма-глутамілтрансферази (GGT) |
|
З боку шкіри та підшкірної тканини |
Дуже часто · Алергічні висипання на шкірі, які часто супроводжуються свербежем · Облисіння Часто · Свербіж · Пітливість Рідко · Тяжкі шкірні реакції, зокрема десквамація, та бульозні висипання на шкірі · Виразки · Пухирцеві формування · Злущування Дуже рідко · Токсичний епідермальний синдром · Синдром Стівенса-Джонсона |
|
З боку опорно-рухового апарату |
Часто · Біль у спині · Міалгія |
|
З боку видільної та сечостатевої системи |
Часто · Гематурія · Помірна протеїнурія Нечасто · Ниркова недостатність
|
|
Загальні розлади |
Дуже часто · Гpипоподібні симптоми, про які найчастіше повідомлялося: пропасниця, головний біль, озноб, міалгія, астенія та відсутність aпeтитy. Кашель, риніт, нездужання, пітливість та розлади сну - симптоми, про які також було повідомлено. · Набряки, зокрема периферичні (у тому числі на обличчі), що зникали із припиненням лікування Часто · Пропасниця · Астенія · Озноб Рідко · Помірні шкірні реакції у місці ін'єкції |
|
Ушкодження, отруєння та ускладнення при проведенні процедури |
Рідко · Радіотоксичність · «Радіаційна пам'ять» |
Комбіноване застосування при раку молочної залози.
Частота випадків явищ гематологічної токсичності ступеня ІІІ та IV, зокрема нейтропенії, підвищується при комбінованому застосуванні гемцитабіну з паклітакселем, хоча підвищення частоти виникнення даних побічних реакцій не асоційоване з підвищеною частотою виникнення інфекцій або геморагічних явищ. Слабкість та фебрильна нейтропенія спостерігаються частіше при комбінованому застосуванні гемцитабіну з паклітакселем. Слабкість, яка не асоційована з анемією, зазвичай проходить після першого циклу терапії.
|
Побічні явища ІІІ та IV ступеня при монотерапії паклітакселем порівняно з комбінованим застосуванням гемцитабіну з паклітакселем |
||||
|
Кількість пацієнтів (%) |
||||
|
Монотерапія паклітакселем (N=3D259) |
Комбіноване застосування гемцитабіну з паклітакселем (N=3D262) |
|||
|
Ступінь ІІІ |
Ступінь IV |
Ступінь ІІІ |
Ступінь IV |
|
|
Лабораторні показники |
||||
|
Анемія |
5 (1,9) |
1 (0,4) |
15 (5,7) |
3 (1,1) |
|
Тромбоцитопенія |
0 |
0 |
14 (5,3) |
1 (0,4) |
|
Нейтропенія |
11 (4,2) |
17 (6,6)* |
82 (31,3) |
45 (17,2)* |
|
Нелабораторні показники |
||||
|
Фебрильна нейтропенія |
3 (1,2) |
0 |
12 (4,6) |
1 (0,4) |
|
Слабкість |
3 (1,2) |
1 (0,4) |
15 (5,7) |
2 (0,8) |
|
Діарея |
5 (1,9) |
0 |
8 (3,1) |
0 |
|
Моторна нейропатія |
2 (0,8) |
0 |
6 (2,3) |
1 (0,4) |
|
Сенсорна нейропатія |
9 (3,5) |
0 |
14 (5,3) |
1 (0,4) |
* Нейтропенія IV ступеня, яка тривала понад 7 днів, спостерігалась у 12,6 % пацієнтів при комбінованому застосуванні та у 5 % пацієнтів при застосуванні тільки паклітакселу.
Комбіноване застосування при раку сечового міхура.
|
Побічні явища ІІІ та IV ступеня при застосуванні МВДЦ (метотрексат, вінбластин, доксорубіцин, цисплатин) порівняно з комбінованим застосуванням гемцитабіну з цисплатином |
||||
|
Кількість пацієнтів (%) |
||||
|
Комбінація МВДЦ (N=3D196) |
Комбіноване застосування гемцитабіну з цисплатином (N=3D200) |
|||
|
Ступінь ІІІ |
Ступінь IV |
Ступінь ІІІ |
Ступінь IV |
|
|
Лабораторні показники |
||||
|
Анемія |
30 (16) |
4 (2) |
47 (24) |
7 (4) |
|
Тромбоцитопенія |
15 (8) |
25 (13) |
57 (29) |
57 (29) |
|
Нелабораторні показники |
||||
|
Нудота та блювання |
37 (19) |
3 (2) |
44 (22) |
0 (0) |
|
Діарея |
15 (8) |
1 (1) |
6 (3) |
0 (0) |
|
Інфекція |
19 (10) |
10 (5) |
4 (2) |
1 (1) |
|
Стоматит |
34 (18) |
8 (4) |
2 (1) |
0 (0) |
Комбіноване застосування при раку яєчників.
|
Побічні явища ІІІ та IV ступеня при монотерапії карбоплатином порівняно з комбінованим застосуванням гемцитабіну з карбоплатином |
||||
|
Кількість пацієнтів (%) |
||||
|
Карбоплатин (N=3D174) |
Комбіноване застосування гемцитабіну з карбоплатином (N=3D175) |
|||
|
Ступінь ІІІ |
Ступінь IV |
Ступінь ІІІ |
Ступінь IV |
|
|
Лабораторні показники |
||||
|
Анемія |
10 (5,7) |
4 (2,3) |
39 (22,3) |
9 (5,1) |
|
Нейтропенія |
19 (10,9) |
2 (1,1) |
73 (41,7) |
50 (28,6) |
|
Тромбоцитопенія |
18 (10,3) |
2 (1,1) |
53 (30,3) |
8 (4,6) |
|
Лейкопенія |
11 (6,3) |
1 (0,6) |
84 (48,0) |
9 (5,1) |
|
Нелабораторні показники |
||||
|
Геморагія |
0 (0) |
0 (0) |
3 (1,8) |
0 (0) |
|
Фебрильна нейтропенія |
0 (0) |
0 (0) |
2 (1,1) |
0 (0) |
|
Інфекція без нейтропенії |
0 (0) |
0 (0) |
0 (0) |
1 (0,6) |
Явище сенсорної нейропатії також спостерігалося частіше при комбінованому застосуванні порівняно із застосуванням одного карбоплатину.
Термін придатності. 2 роки.
Умови зберігання.
Зберігати при температурі не вище 25 °С в оригінальній упаковці.
Зберігати в недоступному для дітей місці. Не заморожувати.
Несумісність
Цей лікарський засіб не слід змішувати з іншими лікарськими засобами.
Упаковка
По 1 г препарату в скляних флаконах, по 1 флакону у картонній упаковці.
Категорія відпуску. За рецептом.
Виробник.
Eмкур Фармасьютікалс Лтд.
Місцезнаходження виробника та адреса місця провадження його діяльності
Плот № П-2, IT Парк, Фаза II MIДС, Хіндіваді, Пуне, ІН 411 057, Індія.
Інші медикаменти цього ж виробника
Форма: капсули тверді по 200 мг № 60 у контейнері № 1
Форма: концентрат для розчину для інфузій, 5 мг/мл по 20 мл (100 мг) у флаконах № 1
Форма: таблетки по 2,5 мг № 30 (10х3) у блістерах
Форма: таблети, вкриті плівковою оболонкою по 50 мг по 30 таблеток у пластиковому контейнері, по 1 пластиковому контейнеру у картонній упаковці
Форма: концентрат для розчину для інфузій, 5 мг/мл по 10 мл (50 мг) у флаконах № 1