Рефакто Аф
Реєстраційний номер: UA/15929/01/02
Імпортер: Пфайзер Ейч. Сі. Пі. Корпорейшн
Країна: СШААдреса імпортера: 235 Іст 42-га Стріт, Нью-Йорк, НЙ 10017-5755, США
Форма
ліофілізат для розчину для ін`єкцій по 500 МО; 1 флакон з ліофілізатом у комплекті з 1 попередньо наповненим шприцом з розчинником по 4 мл (натрію хлорид (9 мг/мл), вода для ін`єкцій), 1 адаптером для флакону, 1 системою для інфузії, 2 тампонами зі спиртом, 1 пластирем та 1 марлевою подушечкою в картонній коробці
Склад
1 флакон містить 500 МО мороктокогу альфа
Виробники препарату «Рефакто Аф»
Країна: Іспанія
Адреса: Аутовіа дель Норте А1, Км 23, десвіо Алгете, Км. 1, сан Себастіан де лос Реєс, 28700 Мадрид, Іспанія
Країна: Іспанія
Адреса: Полігоно Індастріал Ла Міна Авда. Де лос Реєс, Наве59. 28770 Колменар Вієджо Мадрид, Іспанiя
Країна: Німеччина
Адреса: Шутценштрассе 87 та 99-101, Равенсбург, Баден-Вюртенберг, 88212, Німеччина (виробництво розчинника в шприцах та контроль якості (окрім дослідження герметичності, сили тертя поршня), візуальний контроль розчинника);
Інструкція по застосуванню
для медичного застосування лікарського засобу
РеФакто АФ
(ReFacto® AF)
Склад
діюча речовина: фактор коагуляції крові VIII рекомбінантний (мороктоког альфа);
1 флакон містить 250 МО або 500 МО, або 1000 МО, або 2000 МО мороктокогу альфа;
допоміжні речовини: цукроза, кальцію хлориду дигідрат, L-гістидин, полісорбат 80, натрію хлорид.
Розчинник: натрію хлорид (9мг/мл), вода для ін'єкцій.
Лікарська форма
Ліофілізат для розчину для ін'єкцій.
Основні фізико-хімічні властивості: ліофілізат білого кольору, практично вільний від чітко видимих сторонніх часток, вологи та дефектів закупорки флакона;
розчинник: безбарвний прозорий розчин, практично вільний від видимих включень.
Фармакотерапевтична група
Антигеморагічні засоби. Фактор згортання крові VIII. Код АТХ В02В D02.
Фармакологічні властивості
Фармакодинаміка.
РеФакто АФ містить рекомбінантний фактор згортання крові VIII з видаленим В-доменом (мороктоког альфа). Активність препарату в міжнародних одиницях (МО) визначається за допомогою методу хромогенного аналізу згідно Європейської Фармакопеї. Специфічна активність РеФакто АФ становить 7,600-13,800 МО/мг білка.
Мороктоког альфа - це глікопротеїн з приблизною молекулярною масою 170 000 Да, що складається з 1438 амінокислот, послідовність яких подібна до 90 + 80 кДа форми фактора VIII (тобто з видаленим В-доменом), а посттрансляційні модифікації подібні до тих, що наявні у молекулі, отриманій з плазми крові.
Мороктоког альфа виготовляють за допомогою технології рекомбінантної ДНК з використанням клітин яєчників китайських хом'яків. Процес виробництва РеФакто АФ був модифікований таким чином, що виключає можливість потрапляння екзогенного білка людського або тваринного походження під час культивування клітин, очищення або виробництва готового продукту; одночасно назву препарату було змінено з РеФакто на РеФакто АФ.
Функціональні характеристики РеФакто АФ подібні до характеристик ендогенного фактора VIII. Активність фактора VIII значно знижена у пацієнтів із гемофілією А, тому їм необхідна замісна терапія.
При інфузії препарату пацієнтам із гемофілією, введений фактор VIII зв'язується з фактором Віллебранда, що циркулює в крові.
Активований фактор VIII діє як кофактор до активованого фактора ІХ, прискорюючи перетворення фактора Х на активований фактор Х. Активований фактор Х перетворює протромбін на тромбін. Потім тромбін перетворює фібриноген на фібрин і утворюється тромб. Гемофілія А - це зчеплене зі статтю спадкове порушення згортання крові, обумовлене зниженим рівнем фактора VIII:С, яке призводить до профузних крововиливів у суглоби, м'язи або внутрішні органи. Крововиливи можуть виникати спонтанно або в результаті випадкових чи хірургічних травм. При застосуванні замісної терапії рівень фактора VIII в плазмі крові підвищується, що дає змогу тимчасово скоригувати недостатність цього фактора та зменшити схильність до кровотеч.
Індукція імунної толерантності.
Дані стосовно індукції імунної толерантності збиралися серед пацієнтів із гемофілією А, у яких утворилися інгібітори фактора VIII. У рамках базового клінічного дослідження РеФакто із залученням 25 пацієнтів, які раніше не отримували лікування, були зібрані дані щодо індукції імунної толерантності (див. розділ «Побічні реакції»). З 25 цих пацієнтів у 20 пацієнтів титр інгібіторів зменшився до рівня < 0,6 БО (одиниць Бетезда), з яких у 11 з 15 спочатку спостерігалися високі титри (≥ 5 БО), а у 9 з 10 - низькі. Аналогічне зменшення титрів спостерігалося у 5 з 6 пацієнтів, у яких були низькі титри інгібіторів, але вони не отримували лікування індукцією імунної толерантності. Спостереження віддалених наслідків не проводилися.
Фармакокінетика.
Фармакокінетичні властивості РеФакто, визначені у перехресному дослідженні РеФакто та концентрату фактора VIII, отриманого з плазми крові, за участю 18 пацієнтів, які раніше отримували лікування (попередньо ліковані пацієнти - ПЛП), зазначені в таблиці 1. Дослідження кількісного вмісту проводили із застосуванням хромогенного субстрату (див. розділ Спосіб застосування та дози).
Таблиця 1
|
Оцінка фармакокінетичних параметрів РеФакто у ПЛП з гемофілією А |
|||
|
Параметр |
Середнє |
СВ |
Медіана |
|
AUCt (MO·год/мл) |
19,9 |
4,9 |
19,9 |
|
t ½ (год) |
14,8 |
5,6 |
12,7 |
|
CL (мл/год·кг) |
2,4 |
0,75 |
2,3 |
|
середній час утримання (год) |
20,2 |
7,4 |
18,0 |
|
відновлення (МО/дл збільшення фактора VIII:C на МО/кг застосованого фактора VIII) |
2,4 |
0,38 |
2,5 |
Скорочення: AUCt - площа під кривою «концентрація в плазмі - час» від нуля до останньої вимірюваної концентрації; t ½ - період напіввиведення; CL - кліренс; СВ - стандартне відхилення.
У дослідженні, в якому методом хромогенного аналізу порівнювалась активність РеФакто АФ, РеФакто та фактора VIII у плазмі крові пацієнтів, була встановлена біоеквівалентність РеФакто АФ до РеФакто. Співвідношення середніх геометричних значень фармакокінетичних параметрів, визначених методом найменших квадратів, РеФакто АФ до РеФакто становили 100,6 %, 99,5 % та 98,1 % для відновлення, AUCt та AUC∞ (площа під кривою «концентрація в плазмі - час» від нуля до нескінченності) відповідно. Відповідні 90 % довірчі інтервали для співвідношення середніх геометричних значень фармакокінетичних параметрів РеФакто АФ до РеФакто були в межах вікна біоеквівалентності (від 80 до 125 %), що свідчить про біоеквівалентність РеФакто АФ до РеФакто.
У перехресному фармакокінетичному дослідженні фармакокінетичні параметри РеФакто АФ визначали у 25 пацієнтів (≥ 12 років), які раніше отримували лікування, на початку дослідження та через шість місяців багаторазового застосування РеФакто АФ. Співвідношення середніх геометричних значень фармакокінетичних параметрів, визначених методом найменших квадратів, через 6 місяців застосування в порівнянні з вихідними показниками становили 107 %, 100 % та 104 % для відновлення, AUCt та AUC∞ відповідно. Відповідні 90 % довірчі інтервали для співвідношення фармакокінетичних параметрів через 6 місяців дослідження в порівнянні з вихідними показниками були в межах вікна еквівалентності (від 80 % до 125 %). Це свідчить про відсутність залежних від часу змін у фармакокінетичних властивостях РеФакто АФ.
У тому ж самому дослідженні, в якому активність препарату РеФакто АФ, рекомбінантного фактора VIII з повною довжиною молекули (препарат порівняння) та активність фактора VIII визначали у зразках плазми пацієнтів (30 пацієнтів, які раніше отримували лікування ≥ 12 років) за допомогою одноетапного аналізу згортання крові РеФакто АФ продемонстрував фармакокінетичну біоеквівалентність до препарату порівняння при застосуванні стандартного підходу до встановлення біоеквівалентності.
У пацієнтів, які раніше не отримували лікування, фармакокінетичні параметри РеФакто оцінювали за допомогою методу хромогенного аналізу. У цих пацієнтів (n =3D 59; медіана віку 10 ± 8,3 місяці) середнє відновлення на тижні «0» становило 1,5 ± 0,6 МО/дл на МО/кг (у межах 0,2-2,8 МО/дл на МО/кг), що нижче за величини, отримані для пацієнтів, які раніше отримували лікування, на тижні «0» із середнім значенням відновлення 2,4 ± 0,4 МО/дл на МО/кг (у межах 1,1-3,8 МО/дл на МО/кг). У пацієнтів, які раніше не отримували лікування, середнє відновлення було стабільним протягом 2-річного періоду (5 візитів протягом зазначеного часу) та знаходилося в межах 1,5-1,8 МО/дл на МО/кг. Дослідження за допомогою популяційної фармакокінетичної моделі, що включала дані 44 пацієнтів, які раніше не отримували лікування, показало, що середній прогнозований період напіввиведення становить 8,0 ± 2,2 години.
Клінічні характеристики
Показання
Лікування та профілактика кровотеч у пацієнтів із гемофілією А (вроджений дефіцит фактора згортання крові VIII).
РеФакто АФ призначений для застосовування у дорослих та у дітей будь-якого віку, включаючи немовлят.
РеФакто АФ не містить фактор Віллебранда і тому не показаний для лікування хвороби Віллебранда.
Протипоказання
Гіперчутливість до діючої речовини або до будь-якої з допоміжних речовин.
Відома алергічна реакція на білки хом'яка.
Взаємодія з іншими лікарськими засобами та інші види взаємодій
Про взаємодію рекомбінантного фактора коагуляції крові людини VIII з іншими препаратами не повідомлялось.
Особливості застосування
Гіперчутливість
РеФакто АФ може спричинити розвиток реакції гіперчутливості алергічного типу. Препарат містить незначні кількості білків хом'яка. Пацієнтам слід порадити, що при виникненні симптомів гіперчутливості необхідно негайно припинити застосування препарату та звернутися до лікаря. Пацієнтам необхідно повідомити про ранні ознаки розвитку реакцій гіперчутливості, включаючи кропив'янку, генералізовану кропив'янку, відчуття стиснення в грудній клітці, дихання зі свистом, артеріальну гіпотензію та анафілаксію.
У разі виникнення шоку слід здійснювати стандартне медичне лікування шокового стану.
Нейтралізуючі антитіла (інгібітори)
Вироблення нейтралізуючих антитіл (інгібіторів) фактора VIII є відомим ускладненням, що виникає при лікуванні осіб із гемофілією А. Зазвичай це імуноглобуліни IgG, дія яких спрямована проти прокоагулянтної активності фактора VIII. Їх наявність у плазмі визначають методом Бетезда (модифікація Ніймегена) та виражають у БО/мл. Ризик утворення інгібіторів корелює з тривалістю застосування фактора VIII, причому він найвищий протягом перших 20 днів застосування препарату. У рідкісних випадках інгібітори можуть вироблятися після перших 100 днів лікування препаратом.
Випадки повторного утворення інгібіторів (низький титр) спостерігалися при переході з одного препарату рекомбінантного фактора VIII на інший у пацієнтів, які раніше отримували лікування препаратом протягом більше 100 днів та у яких в анамнезі наявне утворення інгібіторів. У зв'язку з цим рекомендується здійснювати ретельний моніторинг усіх пацієнтів щодо вироблення інгібіторів після будь-якої зміни препарату.
Необхідно проводити ретельний моніторинг пацієнтів, які отримують лікування рекомбінантними факторами згортання крові VIII, щодо утворення інгібіторів за допомогою належного клінічного спостереження та лабораторних аналізів. Якщо очікувані рівні активності фактора VIII у плазмі крові не досягаються або якщо кровотеча не зупиняється при застосуванні відповідної дози препарату, слід провести аналізи для визначення наявності інгібіторів фактора VIII. Для пацієнтів з високими титрами інгібіторів терапія фактором VIII може бути неефективною та слід розглянути можливість застосування інших препаратів. Лікування таких пацієнтів повинні проводити лікарі з досвідом лікування гемофілії та випадків вироблення інгібіторів фактора VIII.
Повідомлення про недостатню ефективність
У клінічних дослідженнях та під час постмаркетингового застосування РеФакто повідомлялося про випадки недостатньої ефективності препарату, переважно у пацієнтів, які застосовували препарат з метою профілактики. Відсутність ефективності описувалася як крововиливи в таргетні суглоби, крововиливи в нові суглоби або суб'єктивне відчуття пацієнтом початку нової кровотечі. При призначенні РеФакто АФ важливо індивідуально підбирати дозування та відстежувати рівень фактора в кожного пацієнта для забезпечення адекватної терапевтичної відповіді (див. розділ «Побічні реакції»).
Настійно рекомендується кожного разу при введенні пацієнту препарату РеФакто АФ записувати назву, що зазначена на картонній упаковці, та номер серії лікарського засобу для того, щоб зберегти інформацію про те, яку саме серію препарату було введено певному пацієнту. Пацієнти можуть прикріпити одну з відривних етикеток з флакона або попередньо наповненого шприца, щоб задокументувати номер серії у своєму щоденнику, або для повідомлення про будь-які побічні ефекти.
Серцево-судинні ускладнення
У пацієнтів з наявними факторами ризику розвитку серцево-судинних ускладнень замісне лікування фактором VIII може призвести до підвищення кардіоваскулярних ризиків.
Ускладнення, пов'язані з катетером
У разі необхідності використання пристрою для центрального венозного доступу слід розглянути ризик ускладнень, пов'язаних з його використанням, у тому числі з місцевими інфекціями, бактеріємією та тромбозом катетера (див. розділ «Побічні реакції»).
Вміст натрію
Після розчинення порошку 1 флакон містить 1,23 ммоль (або 29 мг) натрію. Дану інформацію слід враховувати пацієнтам, які дотримуються дієти з обмеженням вживання натрію.
Застосування у період вагітності або годування груддю
Дослідження впливу фактора VIII на репродуктивну функцію тварин не проводилися, тому дані щодо його впливу на фертильність відсутні. Оскільки гемофілія А рідко виникає в жінок, досвід застосування фактора VIII в період вагітності та годування груддю відсутній. Тому фактор VIII слід застосовувати в період вагітності та годування груддю лише за нагальної потреби.
Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами
РеФакто АФ не впливає на здатність керувати транспортними засобами та працювати з іншими механізмами.
Спосіб застосування та дози
Лікування слід розпочинати під наглядом лікаря з досвідом лікування гемофілії А.
Контроль лікування.
Для визначення дози, необхідної протягом курсу лікування, та частоти повторних інфузій рекомендується проводити дослідження рівня фактора VIII. У різних пацієнтів відповідь на введення фактора VIII може варіювати, що проявляється у різних рівнях відновлення та різних періодах напіввиведення. Доза, розрахована за масою тіла пацієнта, може потребувати корегування в хворих з надмірною або недостатньою масою тіла. Зокрема, у випадку значних хірургічних втручань ретельний моніторинг замісної терапії за допомогою аналізу коагуляції (активності фактора VIII у плазмі) є обов'язковим.
Для моніторингу рівня активності фактора VIII у пацієнтів під час лікування препаратом РеФакто АФ рекомендується застосовувати метод хромогенного аналізу.У випадках, коли для визначення активності фактора VIII у зразках крові пацієнтів використовують in vitro активований частковий тромбопластиновий час (АЧТЧ), розрахований за допомогою одноетапного аналізу коагуляційної активності, на результати активності фактора VIII можуть впливати як типи реагентів, так і стандарті зразки, які застосовувались при кількісному визначенні. Також можуть бути суттєві відмінності між результатами кількісного визначення, отриманими з використанням хромогенного методу аналізу та визначенням АЧТЧ шляхом одноетапного аналізу коагуляційної активності. Звичайно результати, отримані методом одноетапного аналізу коагуляційної активності, на 20-50 % нижчі за результати кількісного визначення, виконаного із застосуванням хромогенного методу аналізу. Для корекції цієї розбіжності можна застосовувати лабораторний стандарт РеФакто АФ (див. розділ «Фармакокінетика»). Передусім це важливо, коли використовують інші лабораторії та/або реагенти.
Дозування.
Дозування та тривалість замісної терапії залежать від ступеня недостатності фактора VIII, локалізації та тяжкості кровотечі, а також від клінічного стану пацієнта. Дози, що застосовуються, необхідно коригувати відповідно до клінічної відповіді пацієнта. За наявності інгібіторів може бути потрібне застосування більш високих доз препарату або призначення відповідного специфічного лікування.
Кількість одиниць фактора VIII, що застосовують, зазначають у МО, пов'язаних з діючим стандартом ВООЗ для препаратів, які містять фактор VIII. Активність фактора VIII у плазмі крові зазначається у відсотках (відносно до нормальної людської плазми) або у міжнародних одиницях (відносно міжнародного стандарту для фактора VIII у плазмі). 1 МО активності фактора VIII еквівалентна кількості фактора VIII в 1 мл нормальної людської плазми.
Активність іншого препарату мороктокогу альфа (Ксинта), зареєстрованого поза межами України, визначається за допомогою виробничого стандарту активності, який було відкалібровано відповідно міжнародного стандарту ВООЗ з використанням одноетапного аналізу коагуляційної активності. Через відмінності між методами, які використовуються для визначення активності препаратів Ксинта і РеФакто АФ, 1 МО препарату Ксинта (калібрований за допомогою одноетапного аналізу) приблизно еквівалентна 1,38 МО препарату РеФакто АФ (каліброваного за допомогою хромогенного аналізу). Якщо пацієнту, який зазвичай отримує лікування препаратом Ксинта, призначають РеФакто АФ, лікар може розглянути можливість коригування дози в залежності від показників відновлення рівня фактора VIII.
Пацієнтам із гемофілією А слід порекомендувати мати при собі достатню кількість препарату фактора VIII відповідно до діючої схеми лікування на випадок, якщо лікування буде потрібним під час подорожі. Слід порадити пацієнтам консультуватися із лікарем напередодні подорожі.
Лікування за необхідності.
Розрахунок необхідної дози фактора VIII базується на емпіричних даних про те, що 1 МО фактора VIII на 1 кг маси тіла збільшує активність фактора VIII у плазмі крові на 2 МО/дл. Необхідна доза розраховується за формулою:
необхідна кількість одиниць (МО) =3D
маса тіла (кг) × бажане збільшення кількості фактора VIII (% або МО/дл) × 0,5 (МО/кг на МО/дл),
де 0,5 МО/кг на МО/дл є зворотною величиною відновлення, яке зазвичай спостерігається після інфузії фактора VIII.
Дозу призначеного препарату та частоту застосування завжди слід визначати у кожному випадку окремо, з огляду на клінічну ефективність препарату.
У разі виникнення геморагічних явищ, активність фактора VIII у плазмі крові не повинна падати нижче рівнів (у % або у МО/дл), зазначених у таблиці 2. Цю таблицю можна використовувати як настанову щодо дозування фактора VIII при кровотечах та хірургічному втручанні.
Таблиця 2
|
Тяжкість кровотечі/ Тип хірургічного втручання |
Необхідний рівень фактора VIII (% від нормального або МО/дл плазми) |
Частота введення (години)/ Тривалість терапії (дні) |
|
Кровотеча |
||
|
Початкові ознаки гемартрозу, крововиливів у м'язи або кровотечі в ротовій порожнині |
20-40 |
Введення повторювати кожні 12-24 години щонайменше протягом 1 дня до припинення кровотечі, про що свідчить відсутність болю або наявність загоювання |
|
Гемартрози та м'язові кровотечі середнього ступеня тяжкості або гематоми |
30-60 |
Введення повторювати кожні 12-24 години протягом 3-4 днів або довше до зникнення болю та відновлення рухливості кінцівок |
|
Кровотечі, що загрожують життю |
60-100 |
Введення повторювати кожні 8-24 години до зникнення загрози життю |
Хірургічні втручання |
||
|
Незначні хірургічні втручання, включаючи видалення зуба |
30-60 |
Введення повторювати кожні 24 години, щонайменше протягом 1 дня, до загоювання |
|
Значні хірургічні втручання |
80-100 (до та після хірургічного втручання) |
Введення повторювати кожні 8-24 години до адекватного загоювання рани, потім продовжувати терапію протягом щонайменше 7 днів для підтримання активності фактора VIII на рівні 30-60 % (МО/дл) |
Профілактика
Для довготривалої профілактики кровотеч пацієнтам із тяжкою гемофілією А звичайно застосовують 20-40 МО фактора VIII на кг маси тіла кожні 2-3 дні. У деяких випадках, особливо в пацієнтів молодого віку, може виникнути потреба в збільшенні дози або частоти введення препарату.
Діти
При лікуванні препаратом РеФакто АФ дітей молодшого віку (віком до 6 років) може виникнути необхідність у застосуванні більш високих доз порівняно з тими, що застосовуються дорослим пацієнтам і дітям старшого віку. В дослідженні з використанням фармакокінетичного аналізу за участю дітей віком до 6 років було виявлено, що в цієї категорії пацієнтів період напіввиведення та відновлення менший в порівнянні з показниками, що характерні для дітей старшого віку та дорослих (див. розділ «Фармакокінетика»). Під час клінічних досліджень застосування препарату РеФакто з метою профілактики, діти віком до 6 років отримували у середньому дозу 50 МО/кг, при цьому протягом року спостерігалося в середньому 6,1 епізоду геморагічних явищ. Діти старшого віку та дорослі отримували препарат у середньому дозуванні 27 МО/кг та мали в середньому 10 епізодів геморагічних явищ протягом року. В умовах клінічного дослідження середня доза РеФакто на одну інфузію при епізодах геморагічних явищ у дітей віком до 6 років була вищою за середню дозу, що застосовувалася дітям старшого віку та дорослим (51,3 МО/кг та 29,3 МО/кг відповідно).
Пацієнти літнього віку
Пацієнти віком 65 років і старше не залучались у клінічні дослідження. Загалом, дозу для літніх пацієнтів слід підбирати індивідуально.
Пацієнти з нирковою або печінковою недостатністю
Під час клінічних досліджень не вивчали коригування дози для пацієнтів з нирковою або печінковою недостатністю.
Спосіб застосування.
Внутрішньовенне застосування.
РеФакто АФ вводять шляхом внутрішньовенної інфузії протягом декількох хвилин після розчинення у відповідному розчиннику. Швидкість введення визначається ступенем комфорту пацієнта. Особам, які вводять препарат, але не є медичними працівниками, рекомендується пройти відповідне навчання.
Ліофілізований порошок розчиняють у відповідному розчиннику (розчин натрію хлориду 0,9 %), що додається у попередньо наповненому шприці, використовуючи стерильний адаптер. Після додавання розчинника флакон слід обережно повертати до повного розчинення порошку.
Більш детальна інформація щодо приготування та застосування розчину наведена нижче.
Препарат РеФакто АФ після розчинення містить полісорбат 80, який має здатність прискорювати екстракцію ди-(2-етилгексил)фталату з полівінілхлориду (ПВХ). Дану особливість необхідно враховувати при приготуванні та застосуванні препарату, включаючи час зберігання у ПВХ-контейнері після приготування розчину. Важливо точно дотримуватися рекомендацій, наданих у розділі «Умови зберігання».
Після розчинення отриманий розчин набирають назад у шприц. Розчин повинен бути прозорим або злегка опалесцентним та безбарвним. Розчин слід викинути, якщо спостерігаються включення чужорідних часточок або зміна кольору.
Невикористані залишки препарату або матеріали, що використовувалися для розчинення та введення препарату, мають бути утилізовані згідно з місцевими вимогами.
Приготування розчину
1. Доведіть температуру ліофілізату та розчинника у попередньо наповненому шприці до кімнатної температури.
2. Зніміть пластикову flip-top кришку з флакона РеФакто АФ, щоб відкрити центральну частину гумової пробки.
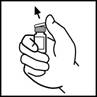
3. Протріть верхню частину флакона тампоном зі спиртом, що додається до упаковки, або використайте інший антисептичний розчин та дайте висохнути. Після очищення не торкайтеся руками гумової пробки та не допускайте, щоб вона торкалася будь-яких поверхонь.
4. Відігніть захисне покриття прозорої пластикової упаковки адаптера до флакона. Не виймайте адаптер з упаковки.
5. Поставте флакон на рівну поверхню. Тримаючи упаковку з адаптером, насадіть адаптер на флакон. Сильно натисніть так, щоб адаптер з клацанням зайняв своє місце на верхній частині флакона, а вістря адаптера пройшло крізь пробку флакона.
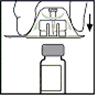
6. Зніміть упаковку з адаптера та утилізуйте її.
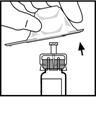
7. Приєднайте шток поршня до шприца з розчинником: вставте шток в отвір у стопері шприца, сильно натискайте та прокручуйте шток, поки він не ввійде надійно у стопер.
8. Відламайте пластиковий ковпачок-насадку з індикатором першого відкриття зі шприца з розчинником, порушивши цілісність перфорації на ковпачку. Це роблять, розхитуючи ковпачок доти, доки перфорація не буде зламана. Не торкайтеся внутрішньої поверхні ковпачка або кінчика шприца. Може виникнути необхідність вдягнути ковпачок назад (якщо приготований РеФакто АФ не буде використаний негайно), тому відкладіть його, поставивши отвором догори.
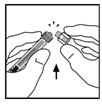
9. Поставте флакон на рівну поверхню. Приєднайте шприц з розчинником до адаптера флакона, вставляючи наконечник шприца у отвір адаптера, сильно натискаючи та прокручуючи шприц за ходом годинника до отримання надійного з'єднання.
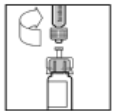
10. Повільно натисніть на шток поршня, щоб ввести весь розчинник у флакон РеФакто АФ.
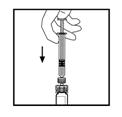
11. Залишаючи шприц приєднаним до адаптера, обережно обертайте флакон до повного розчинення порошку.

12. Перед застосуванням приготований розчин слід візуально перевірити щодо наявності сторонніх часток. Отриманий розчин повинен бути прозорим або злегка опалесцентним та безбарвним.
Якщо використовується більше одного флакона препарату РеФакто АФ на інфузію, кожний флакон слід готувати згідно з попередніми інструкціями. Далі шприц для розчинника слід видалити, залишаючи адаптер до флакона на місці, а для відбору розчину з кожного флакона, можна використовувати один великий шприц з замком Люера.
13. Впевнившись, що поршень залишається повністю введеним в циліндр шприца, переверніть флакон. Повільно витягніть увесь розчин через адаптер флакона у шприц.
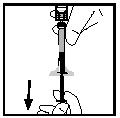
14. Від'єднайте шприц від адаптера флакона, обережно відтягуючи та прокручуючи шприц проти ходу годинника. Викиньте флакон з приєднаним адаптером.
Якщо розчин не буде використано негайно, ковпачок шприцу слід обережно повернути на місце. Не слід торкатися кінчика шприца або внутрішньої поверхні ковпачка.
Приготований розчин слід використати негайно або протягом 3 годин після приготування. До застосування приготований розчин можна зберігати при температурі не вище 25 °С.
Застосування (внутрішньовенна ін'єкція)
РеФакто АФ слід вводити, використовуючи інфузійний набір, що додається, та попередньо наповнений розчинником шприц, що додається, або один стерильний одноразовий пластиковий шприц з замком Люера.
1. Приєднайте шприц до кріплення Люера катетера інфузійного набору.
2. Накладіть джгут та підготуйте ділянку ін'єкції, добре протерши шкіру тампоном зі спиртом, що надається з комплектом.

3. Введіть голку катетера інфузійного набору у вену та зніміть джгут. Видаліть повітря з катетера інфузійного набору, відтягуючи шприц. Приготований препарат слід вводити внутрішньовенно протягом кількох хвилин. Лікар може змінювати рекомендовану швидкість інфузії, щоб процедура інфузії була більш комфортною для Вас.

Слід утилізувати невикористаний розчин, порожній флакон(и) та використані голки та шприци у відповідному контейнері для утилізації медичних відходів, оскільки ці матеріали можуть зашкодити іншим, якщо не будуть утилізовані належним чином.
Діти
РеФакто АФ можна застосовувати дітям будь-якого віку, включаючи немовлят, як зазначено в розділі «Спосіб застосування та дози».
Передозування.
Не повідомлялось про будь-які симптоми передозування при застосуванні препаратів рекомбінантного фактора VIII.
Побічні реакції
Резюме профілю безпеки
Гіперчутливість та алергічні реакції (які можуть включати ангіоневротичний набряк, печіння та поколювання в місці інфузії, озноб, припливи, генералізовану кропив'янку, головний біль, кропив'янку, артеріальну гіпотензію, летаргію, нудоту, збудженість, тахікардію, стиснення в грудях, шум у вухах, блювання, дихання зі свистом) при застосуванні РеФакто АФ виникають рідко та в деяких випадках прогресують до тяжкої форми анафілаксії, включаючи шок (див. розділ «Особливості застосування»)
РеФакто АФ може містити слідові кількості білка хом'яка. Дуже рідко спостерігалося утворення антитіл до білка хом'яка, але клінічні ускладнення були відсутні. В дослідженні РеФакто у 20 з 113 (18 %) пацієнтів, які раніше отримували лікування, спостерігалося підвищення титру антитіл до білка яєчників китайського хом'яка без видимого клінічного ефекту.
Відомо, що при лікуванні пацієнтів із гемофілією А утворюються нейтралізуючі антитіла (інгібітори) до фактора VIII. При застосуванні будь-яких препаратів фактора згортання крові VIII необхідно проводити контроль утворення інгібіторів методом Бетезда (модифікація Ніймегена), зазначаючи їх кількість у БО. Утворення інгібіторів може проявлятися у вигляді недостатньої клінічної відповіді. В таких випадках рекомендується звернутися до спеціалізованого центру з лікування гемофілії.
Перелік побічних реакцій
Інформація, що наведена нижче, відповідає системно-органній класифікації згідно з медичним словником для регуляторної діяльності (MedDRA). Побічні реакції класифіковано відповідно до частоти їх виникнення: дуже часто (≥ 1/10); часто (від ≥ 1/100 до < 1/10) та нечасто (від ≥ 1/1000 до < 1/100). Нижче представлені побічні реакції, що спостерігалися в клінічних дослідженнях РеФакто або РеФакто АФ. Частота зазначена з урахуванням всіх випадкових побічних реакцій, що виникли під час застосування препарату у ході всіх клінічних досліджень із залученням 655 пацієнтів (554 пацієнти, які раніше отримували лікування, та 101 пацієнт, які раніше не отримували лікування).
У кожній групі побічні ефекти зазначені в порядку зменшення їх серйозності.
З боку кровотворної та лімфатичної систем:
дуже часто: інгібування фактора VIII (у пацієнтів, які раніше не отримували лікування);
часто: інгібування фактора VIII (у пацієнтів, які раніше отримували лікування).
З боку імунної системи:
нечасто: анафілактичні реакції.
Метаболічні порушення та розлади харчування:
часто: зниження апетиту.
З боку нервової системи:
дуже часто: головний біль;
часто: запаморочення;
нечасто: периферійна нейропатія, сонливість, дисгевзія.
З боку серця:
нечасто: стенокардія, тахікардія, відчуття серцебиття.
З боку судин:
часто: кровотеча/гематома;
нечасто: гіпотензія, тромбофлебіт, гіперемія обличчя.
З боку дихальної системи, органів грудної клітки та середостіння:
дуже часто: кашель;
нечасто: задишка.
З боку травної системи:
часто: діарея, нудота, біль у животі, блювання.
З боку шкіри та підшкірної тканини:
часто: кропив'янка, свербіж, висипання;
нечасто: гіпергідроз.
З боку скелетно-м'язової системи та сполучної тканини:
дуже часто: артралгія;
часто: міалгія.
Загальні розлади та реакції у місці введення:
дуже часто: підвищення температури тіла;
часто: озноб, ускладнення з боку постійного венозного катетера;
нечасто: астенія, біль, запалення та інші реакції у місці введення.
Лабораторні аналізи:
часто: позитивний тест на антитіла; позитивний тест на антитіла до фактора VIII;
нечасто: підвищення рівня аспартатамінотрасферази, аланінамінотрансферази, білірубіну крові, креатинінфосфокінази крові.
Опис окремих побічних реакцій
Пригнічення фактора VIII.
У клінічному дослідженні застосування РеФакто АФ частота утворення інгібіторів фактора VIII була первинною кінцевою точкою оцінки безпеки у пацієнтів, які раніше отримували лікування.
У ході дослідження за участю 94 пацієнтів повідомлялося про виникнення 2 клінічно безсимптомних випадків тимчасового утворення інгібіторів із низьким титром при медіані експозиції 76 днів (в межах 1-92 днів), що відповідає 2,2 % з 89 пацієнтів при щонайменше 50 днях експозиції. У додатковому дослідженні РеФакто АФ, в якому брали участь 110 пацієнтів, спостерігався 1 de novo випадок та 2 випадки повторного утворення інгібіторів (у всіх випадках із низьким титром, за результатами центральної лабораторії); медіана експозиції становила 58 днів (у межах 5-140 днів) та для 98 пацієнтів експозиція РеФакто АФ становила щонайменше 50 днів. 98 пацієнтів зі 110 продовжили лікування у другому додатковому дослідженні та мали подовжену експозицію РеФакто АФ, медіана якої становила 169 додаткових днів експозиції (у межах 9-425 днів). При цьому спостерігався 1 додатковий de novo випадок утворення інгібіторів із низьким титром. Частота утворення інгібіторів, яка спостерігалася в цих дослідженнях, знаходиться в очікуваних межах.
В клінічному дослідженні із залученням пацієнтів з гемофілією A, які раніше отримували лікування (фактор VIII:C ≤ 2 %), та яким була виконана серйозна хірургічна операція, був зареєстрований 1 випадок вироблення інгібіторів серед 30 пацієнтів, які отримували лікування Рефакто АФ.
У клінічному дослідженні застосування РеФакто із залученням 113 пацієнтів, які раніше отримували лікування, був відмічений 1 випадок утворення інгібіторів. Також були зареєстровані спонтанні постреєстраційні повідомлення про вироблення інгібіторів у високих титрах, у т.ч.у пацієнтів, які раніше отримували лікування.
Клінічні дослідження РеФакто АФ за участю пацієнтів, які раніше не отримували лікування все ще тривають. У клінічному дослідженні РеФакто у 32 з 101 (32 %) пацієнтів, які раніше не отримували лікування (фактор VIII:C < 2 %) спостерігалося утворення інгібіторів. Із 62 пацієнтів із фактором VIII:С < 1 % у 19 пацієнтів спостерігалося утворення інгібіторів (31 %). Із 32 випадків вироблення інгібіторів (у групі з 101 пацієнта) 16 пацієнтів (16 %) було класифіковано як пацієнти з високими титрами (≥ 5 БО) і 16 (16 %) - з низькими титрами (< 5 БО). Медіана кількості днів експозиції до розвитку інгібіторів у цих 32 пацієнтів становила 12 (діапазон 3-49). Із 16 пацієнтів із високими титрами у 15 були проведені заходи з індукції імунної толерантності. Із 16 пацієнтів із низькими титрами заходи з індукції імунної толерантності розпочалася у 10 пацієнтів.
Діти
Повідомлялося про один випадок розвитку кісти в 11-річного пацієнта та один випадок затьмарення свідомості в 13-річного пацієнта; розвиток цих станів може бути пов'язаний із лікуванням препаратом РеФакто АФ.
Безпеку застосування РеФакто АФ оцінювали у дітей та підлітків, які раніше отримували лікування (n =3D 18, віком 12-16 років у дослідженні та n =3D 49, віком 7-16 років у додатковому дослідженні). Незважаючи на те, що дослідження проводили із залученням обмеженої кількості дітей, було встановлено тенденцію до збільшення частоти небажаних явищ у дітей віком 7-16 років порівняно з дорослими.
Дослідження застосування Рефакто АФ дітям віком до 6 років тривають.
Звітування про підозрювані побічні реакції.
Важливо надавати інформацію про підозрювані побічні реакції після реєстрації лікарського засобу. Це дає змогу і далі контролювати співвідношення «користь/ризик» для лікарського засобу. Медичні працівники повинні повідомляти про всі підозрювані побічні реакції згідно з місцевими вимогами.
Термін придатності
Термін придатності ліофілізату 3 роки.
Термін придатності розчинника 5 років.
Умови зберігання
Зберігати в оригінальній упаковці при температурі від 2 до 8 °С. Не заморожувати, щоб уникнути пошкодження попередньо наповненого шприца.
Препарат у межах терміну придатності можна зберігати при температурі не вище 25 °С протягом 3 місяців. Препарат не можна повертати до холодильника, якщо його зберігали при кімнатній температурі.
Приготований розчин слід використати одразу або протягом 3 годин після приготування розчину за умови зберігання при температурі не вище 25 °С.
Зберігати в недоступному для дітей місці.
Несумісність
Оскільки дослідження сумісності цього препарату не проводилися, препарат не можна змішувати з іншими лікарськими засобами, включаючи інші інфузійні розчини.
Для введення розчину необхідно використовувати набір для інфузій, що входить у комплект, оскільки фактор згортання крові VIII може адсорбуватися на внутрішніх поверхнях іншого інфузійного обладнання.
Упаковка
1 флакон з ліофілізатом, 1 попередньо наповнений шприц з розчинником, 1 адаптер для флакону, 1 систему для інфузії, 2 тампони зі спиртом, 1 пластир, 1 марлеву подушечку вкладають в картонну коробку.
Категорія відпуску. За рецептом.
Виробник.
Ваєт Фарма С. А. / Wyeth Farma S.A.
Місцезнаходження виробника та його адреса місця провадження діяльності.
Аутовіа дель Норте А1, Км 23, десвіо Алгете, Км. 1, Сан Себастіан де лос Реєс, 28700 Мадрид, Іспанія / Autovia del Norte A1, Km 23, desvio Algete, Km. 1, San Sebastian de los Reyes, 28700 Madrid, Spain.
Інші медикаменти цього ж виробника
Форма: капсули по 250 мг по 60 капсул у флаконах; по 10 капсул у блістері; по 1 або по 6 блістерів у картонній коробці
Форма: таблетки, вкриті плівковою оболонкою, по 1 мг по 14 таблеток у блістері; по 2 або по 4 блістери в картонній коробці
Форма: таблетки по 0,45 мг/20 мг, по 15 таблеток у блістері; по 2 блістери у саше; по 1 саше у картонній коробці
Форма: порошок та розчинник для розчину для ін`єкцій, 20 мг, по 1 флакону разом з 1 ампулою розчинника в картонній упаковці
Форма: розчин для ін'єкцій або інфузій по 30 млн ОД (300 мкг)/0,5 мл, по 0,5 мл у шприці (І класу); по 1, 5 або 10 попередньо наповнених шприцах місткістю 1 мл у блістері в картонній пачці