Сигніфор Лар
Реєстраційний номер: UA/15926/01/01
Імпортер: Новартіс Фарма АГ
Країна: ШвейцаріяАдреса імпортера: Ліхтштрассе, 35, 4056 Базель, Швейцарія
Форма
порошок для суспензії для ін`єкцій по 20 мг, 1 флакон з порошком у комплекті з розчинником по 2 мл у попередньо заповненому шприці (кармелоза натрію, маніт (E421), полоксамер 188, вода для ін’єкцій), 1 голкою та 1 адаптером в картонній коробці
Склад
1 флакон з порошком для суспензії для ін’єкцій містить 20 мг пасиреотиду (у формі пасиреотиду памоату)
Виробники препарату «Сигніфор Лар»
Країна: Швейцарія
Адреса: Шаффхаусерштрассе, 4332 Штейн, Швейцарія
Країна: Нідерланди
Адреса: Веєрвег 12, 8121 АА Ольст, Нідерланди
Країна: Австрія
Адреса: Біохеміштрассе 10, 6336 Лангкампфен, Австрія (контроль якості за всіма параметрами за виключенням тесту "Бактеріальні ендотоксини", первинне пакування порошку);
Країна: Швейцарія
Адреса: Ліхтштрассе 35, 4056 Базель, Швейцарія
Країна: Німеччина / Швейцарія
Адреса: Берінгштрассе 6-8, 82152 Планегг, Німеччина / Хогенвейдштрассе 6, 4658 Денікен, Швейцарія
Інструкція по застосуванню
для медичного застосування лікарського засобу
СИГНІФОР ЛАР
(SIGNIFOR® LAR®)
Склад
діюча речовина: pasireotide;
1 флакон з порошком для суспензії для ін'єкцій містить 20 мг, 40 мг або 60 мг пасиреотиду (у формі пасиреотиду памоату).
допоміжні речовини:
порошок: полі (D,L-лактид-ко-гліколід) (50-60:40-50), полі (D,L-лактид-ко-гліколід) (50:50);
розчинник: 1 попередньо-заповнений шприц з розчинником по 2 мл містить: кармелозу натрію, маніт (E421), полоксамер 188, воду для ін'єкцій.
Лікарська форма. Порошок для суспензії для ін'єкцій.
Основні фізико-хімічні властивості:
порошок: порошок від злегка жовтуватого до жовтуватого кольору;
розчинник: прозорий розчин від безбарвного до жовтуватого або світло-коричневого кольору.
Фармакотерапевтична група. Гіпофізарні, гіпоталамічні гормони та їх аналоги.
Код АТХ H01C B05.
Фармакологічні властивості.
Фармакодинаміка.
Механізм дії. Пасиреотид - циклогексапептид, ін'єкційний аналог соматостатину. Подібно до природних пептидних гормонів соматостатину-14 та соматостатину-28 (також відомих як фактор, що гальмує виділення соматотропіну [SRIF]) та інших аналогів соматостатину, пасиреотид реалізує свою фармакологічну дію шляхом зв'язування з рецепторами соматостатину. Відомі п'ять підтипів людських рецепторів соматостатину: hsst1, 2, 3, 4 та 5. За нормальних фізіологічних умов ці підтипи рецепторів експресуються в різних тканинах. Аналоги соматостатину з різною потужністю зв'язуються з рецепторами hsst (див. таблицю 1). Пасиреотид з високою афінністю зв'язується з чотирма із п'яти рецепторів hsst.
Таблиця 1. Афінність зв'язування соматостатину (SRIF-14), пасиреотиду, октреотиду та ланреотиду з п'ятьма людськими підтипами рецепторів sst (hsst1-5).
Препарат |
hsst1 |
hsst2 |
hsst3 |
hsst4 |
hsst5 |
|
Соматостатин (SRIF-14) |
0,93 ± 0,12 |
0,15 ± 0,02 |
0,56 ± 0,17 |
1,5 ± 0,4 |
0,29 ± 0,04 |
|
Пасиреотид |
9,3 ± 0,1 |
1,0 ± 0,1 |
1,5 ± 0,3 |
>100 |
0,16 ± 0,01 |
|
Октреотид |
280 ± 80 |
0,38 ± 0,08 |
7,1 ± 1,4 |
>1000 |
6,3 ± 1,0 |
|
Лантреотид |
180 ± 20 |
0,54 ± 0,08 |
14 ± 9 |
230 ± 40 |
17 ± 5 |
Результати представлені у вигляді середнього значення ± середнє значення стандартної похибки (ССП) IC50, вираженого в нмоль/л.
Фармакодинамічні ефекти. Рецептори соматостатину експресуються у багатьох тканинах, особливо у нейроендокринних пухлинах, при яких спостерігається надмірна секреція гормонів, в тому числі гормону росту (ГР) при акромегалії.
Завдяки своєму широкому профілю зв'язування з рецепторами соматостатина пасиреотид здатний стимулювати рецептори підтипу hsst2 та hsst5, необхідні для інгібування секреції ГР та інсуліноподібного фактору росту (ІФР-1), тому він може бути ефективним для лікування акромегалії.
Метаболізм глюкози. В ході рандомізованого подвійного сліпого дослідження за участю здорових добровольців розвиток гіперглікемії при підшкірному застосуванні пасиреотиду в дозах 0,6 мг та 0,9 мг двічі на добу був пов'язаний із значним зменшенням секреції гормонів інсуліну, а також інкретину (а саме, глюкагоноподібного пептиду-1 (ГПП-1) та глюкозозалежного інсулінотропного поліпептиду (ГІП)). Пасиреотид не впливав на чутливість до інсуліну.
Педіатрична популяція. Європейське агентство з лікарських засобів відмовилося від обов'язку подання результатів досліджень препарату Сигніфор у дітей всіх підкатегорій при акромегалії та гіпофізарному гігантизмі (інформацію про застосування у дітей див. у розділі 4.2).
Фармакокінетика.
Поглинання. Відносна біодоступність пасиреотиду при внутрішньом'язовому застосуванні порівняно з підшкірним застосування є повною. Досліджень з метою оцінки біодоступності пасиреотиду у людей не проводилось.
Розподіл. У здорових добровольців пасиреотид при внутрішньом'язовому застосуванні широко розподілявся з великим об'ємом розподілу (Vz/F > 100 літрів). Розподіл між клітинами крові та плазмою не залежить від концентрації і показує, що пасиреотид локалізується переважно у плазмі крові (91 %). Зв'язування з білками плазми є помірним (88 %) і не залежить від концентрації.
На основі даних досліджень in vitro пасиреотид є субстратом ефлюксного транспортера Р-глікопротеїна. На основі даних досліджень in vitro пасиреотид не є субстратом ефлюксного транспортера BCRP (протеїн резистентності раку молочної залози) та інфлюксних транспортерів ОСТ1 (транспортер органічних катіонів 1), ОАТР (поліпептид, що транспортує органічні аніони) 1B1, 1B3 або 2B1. У терапевтичних дозах пасиреотид також не є інгібітором UGT1A1, OATP1B1 або 1B3, OAT1 або OAT3, OCT1 або OCT2, P-gp, BCRP, MRP2 та BSEP.
Біотрансформація. Пасиреотид є високостабільним у метаболічному відношенні. Дані досліджень in vitro свідчать, що пасиреотид не є субстратом, інгібітором або індуктором будь-яких основних ферментів CYP450. У здорових добровольців пасиреотид переважно виявляється у незміненому вигляді у плазмі крові, сечі та калі.
Виведення. Пасиреотид виводиться переважно шляхом печінкового кліренсу (біліарна екскреція) за незначною участю ниркового шляху виведення. У дослідженні ADME (всмоктування, розподіл, метаболізм, виведення) у людей 55,9 ± 6,63 % радіоактивної дози відновлювалось протягом перших 10 днів після введення, включаючи 48,3 ± 8,16 % радіоактивності у калі та 7,63 ± 2,03 % у сечі.
Кліренс (CL/F) внутрішньом'язового пасиреотиду у здорових добровольців становить у середньому 4,5-8,5 л/год.
Лінійність та залежність від часу. Фармакокінетичний стан спокою при внутрішньом'язовому застосуванні пасиреотиду досягається через 3 місяці. Після декількох щомісячних доз пасиреотид при внутрішньом'язовому застосуванні демонструє приблизно дозопропорційний фармакокінетичний вплив у діапазоні доз від 20 мг до 60 мг кожні 4 тижні у пацієнтів з акромегалією.
Особливі категорії пацієнтів. Педіатрична популяція. Дослідження у дітей не проводились.
Пацієнти з порушенням функції нирок. Нирковий кліренс відіграє мінімальну роль у виведенні пасиреотиду у людей. У клінічному дослідженні одноразова підшкірна доза пасиреотиду 900 мкг у пацієнтів з нирковою дисфункцією, легким, помірним та тяжким порушенням функції нирок або хворобою нирок у термінальній стадії (ESRD) не чинила значущого впливу на загальну плазмову експозицію пасиреотиду. Плазмова експозиція незв'язаного пасиреотиду (AUCinf,u) збільшилась у пацієнтів з порушенням функції нирок (легкий: 33%; помірний: 25%, тяжкий: 99%, ESRD: 143%) порівняно з пацієнтами в контрольній групі.
Пацієнти з порушенням функції печінки. Клінічні дослідження внутрішньом'язового застосування пасиреотиду у пацієнтів з порушеною функцією печінки не проводились. У клінічному дослідженні одноразової підшкірної дози пасиреотиду за участю пацієнтів з порушенням функції печінки статистично значущу відмінність було виявлено у пацієнтів з помірним та тяжким порушенням функції печінки (клас В та С за Чайлд-П'ю). У пацієнтів з помірним та тяжким порушенням функції печінки AUC inf зростала на 60 % та 79 %, Cmax - на 67 % та 69 %, а CL/F знижувався на 37 % та 44 % відповідно.
Пацієнти літнього віку (≥ 65 років). Вік не є істотним параметром при популяційному фармакокінетичному аналізі пацієнтів з акромегалією.
Демографічні характеристики. Популяційний фармакокінетичний аналіз внутрішньом'язового застосування пасиреотиду свідчить, що расова належність та стать не впливають на фармакокінетичні показники препарату. Маса тіла незначно впливала на фармакокінетику у дослідженні за участю пацієнтів, які раніше не отримували лікування, але не впливала у дослідженні за участю пацієнтів, яким не забезпечувався достатній контроль захворювання. Жінки з акромегалією піддавались більшому впливу (32 % та 51 %) порівняно з чоловіками у дослідженнях пацієнтів, які не отримували лікування, та пацієнтів, яким не забезпечувався достатній контроль, відповідно; ці відмінності не були клінічно релевантними на основі даних про ефективність та безпеку.
Клінічні характеристики
Показання
Лікування дорослих пацієнтів з акромегалією, у яких хірургічне втручання не є оптимальним або було невдалим, і яким не забезпечується належний контроль за допомогою іншого аналогу соматостатину.
Протипоказання
Підвищена чутливість до активної речовини або до будь-якого компонента препарату. Тяжке порушення функції печінки (клас C за Чайлд-П'ю).
Взаємодія з іншими лікарськими засобами та інші види взаємодій
Очікувані фармакокінетичні взаємодії, які зумовлюють вплив на пасиреотид. Вплив інгібітору Р-глікопротеїна верапамілу на фармакокінетику пасиреотиду у разі підшкірного застосування був випробуваний у дослідженні взаємодії лікарських засобів за участю здорових добровольців. Жодних змін фармакокінетики (швидкості або ступеню впливу) пасиреотиду не спостерігалось.
Очікувані фармакокінетичні взаємодії, що зумовлюють вплив на інші лікарські засоби. Пасиреотид може знижувати відносну біодоступність циклоспорину. При одночасному застосуванні пасиреотиду та циклоспорину може потребуватися корекція дози циклоспорину з метою підтримання терапевтичної концентрації.
Очікувані фармакодинамічні взаємодії. Лікарські засоби, що пролонгують інтервал QT.
Пасиреотид слід застосовувати з обережністю пацієнтам, які одночасно приймають лікарські засоби, що пролонгують інтервал QT, такими як антиаритмічні засоби класу Іа (наприклад хінідин, прокаїнамід, дизопірамід), антиаритмічні засоби класу ІІІ (наприклад аміодарон, дронедарон, соталол, дофетилід, ібутилід), деякі антибактеріальні засоби (еритроміцин внутрішньовенно, ін'єкції пентамідину, кларитроміцин, моксифлоксацин), деякі антипсихотичні засоби (наприклад хлорпромазин, тіоридазин, флуфеназин, пімозид, галоперидол, тіаприд, амісульприд, сертиндол, метадон), деякі антигістамінні засоби (наприклад терфенадин, астемізол, мізоластин), антималярійні засоби (наприклад хлорохін, галофантрин, люмефантрин), деякі протигрибкові засоби (кетоконазол, крім шампуню).
Лікарські засоби, що спричиняють брадикардію. Клінічний моніторинг серцевого ритму, особливо на початку лікування, рекомендується пацієнтам, які отримують пасиреотид одночасно з лікарськими засобами, що спричиняють брадикардію, такими як бета-блокатори (наприклад метопролол, картеолол, пропранолол, соталол), інгібітори ацетилхолінестерази (наприклад, ривастигмін, фазостигмін), деякі блокатори кальцієвих каналів (наприклад верапаміл, дилтіазем, бепридил), деякі антиаритмічні засоби.
Інсулін та протидіабетичні лікарські засоби. Корекція дози (зниження або підвищення) інсуліну та протидіабетичних лікарських засобів (наприклад метформіну, ліраглутиду, вілдагліптину, натеглініду) може потребуватися у разі одночасного застосування з пасиреотидом.
Особливості застосування
Розлади обміну глюкози. Зміни в рівнях глюкози в крові часто відзначалися у здорових добровольців і пацієнтів після лікування пасиреотидом. Гіперглікемія і (рідше) гіпоглікемія спостерігались у пацієнтів, які брали участь у клінічних дослідженнях пасиреотиду.
Ступінь і частота гіперглікемії, що спостерігалась у двох основних дослідженнях за участю пацієнтів з акромегалією, була вище у разі внутрішньом'язового застосування препарату Сигніфор ЛАР, ніж у разі активного контролю (октреотиду внутрішньом'язово або ланреотид у вигляді глибокої підшкірної ін'єкції). В узагальненому аналізі двох основних досліджень загальна частота несприятливих побічних реакцій пов'язаних з гіперглікемією, була 58,6 % (всі ступені) і 9,9 % (критерії загальної токсичності (КЗТ) ступеню 3 і 4) для внутрішньом'язового застосування препарату Сигніфор порівняно з 18,0 % (всі ступені) і 1,1 % (КЗТ ступеню 3 і 4) для активного контролю. У базовому дослідженні пацієнтів, яким не забезпечується достатній контроль за допомогою іншого аналогу соматостатину, частка пацієнтів, які раніше не отримували протидіабетичних засобів, які потребували протидіабетичної терапії в ході дослідження, становила 17,5 % і 16,1 % в групах препарату Сигніфор ЛАР 40 мг і 60 мг в порівнянні з 1,5 % в групі активного контролю; у базовому дослідженні за участю пацієнтів, які не отримували попереднє лікування, частка пацієнтів, які потребували протидіабетичної терапії в ході дослідження, становила 36 % в групі препарату Сигніфор ЛАР в порівнянні з 4,4 % в групі активного контролю.
Пацієнти з акромегалією, у яких розвивалась гіперглікемія, в цілому реагували на протидіабетичну терапію. Зниження дози або припинення лікування пасиреотидом через гіперглікемію були нечастими під час клінічних досліджень пасиреотиду.
Розвиток гіперглікемії пов'язаний зі зменшенням секреції інсуліну (особливо у період після прийому дози) та інкретинових гормонів (а саме, глюкагоноподібного пептиду-1 (ГПП-1) та глюкозозалежного інсулінотропного поліпептиду (ГІП)).
Стан глікемії (глюкоза плазми крові натщесерце/гемоглобін А1с (ГПН/НА1с)) слід оцінити до початку лікування пасиреотидом. Моніторинг ГПН/А1с протягом лікування слід проводити згідно наявних рекомендацій. Самомоніторинг глюкози крові та/або оцінку ГПН слід виконувати кожен тиждень протягом перших 2-3 місяців та потім періодично, відповідно до клінічної практики. Крім того, слід виконати моніторинг ГПН через 4 тижні та гемоглобіну А1с через 3 місяці після закінчення лікування.
Якщо у пацієнта на тлі прийому препарату Сигніфор ЛАР розвивається гіперглікемія, рекомендується розпочати або відкоригувати протидіабетичне лікування згідно з наявними рекомендаціями для контролю гіперглікемії. Якщо неконтрольована гіперглікемія триває, незважаючи на відповідне медикаментозне лікування, слід зменшити дозу препарату Сигніфор ЛАР або припинити лікування.
Пацієнти з поганим глікемічним контролем (визначеним за значенням HbA1c > 8 % на тлі протидіабетичної терапії) можуть мати підвищений ризик розвитку тяжкої гіперглікемії та супутніх ускладнень. У пацієнтів з поганим глікемічним контролем контроль діабету та моніторинг слід посилити до початку та протягом терапії пасиреотидом.
Печінкові тести. У пацієнтів, які приймали пасиреотид, зазвичай спостерігалось незначне транзиторне підвищення рівня амінотрансфераз. Також спостерігалися рідкісні випадки одночасного підвищення АЛТ вище 3 х ВМН (верхня межа норми) та білірубіну вище 2 х ВМН (верхня межа норми). Рекомендується моніторинг функції печінки до початку лікування пасиреотидом та після одного, двох, чотирьох та дванадцяти тижнів лікування, що не супроводжувалось підвищенням рівня загального білірубіну в сироватці крові. Надалі моніторинг функції печінки слід виконувати за клінічними показаннями.
Пацієнтам, у яких розвинулося підвищення рівня трансаміназ, слід провести повторне визначення функції печінки для підтвердження одержаних результатів. Якщо результати підтверджені, то такі пацієнти підлягають частому моніторингу функції печінки до повернення значень до рівнів, які були до лікування. Терапію пасиреотидом слід припинити, якщо у пацієнта розвинулася жовтяниця або інші ознаки клінічно значущої дисфункції печінки, у випадку стійкого підвищення АСТ або АЛТ 5 х ВМН або вище, або АЛТ чи АСТ вище АЛТ 3 х ВМН одночасно з підвищенням білірубіну вище АЛТ 2 х ВМН. Після припинення терапії пасиреотидом слід проводити моніторинг пацієнтів до зникнення симптомів. Лікування не слід поновлювати, якщо є підозра на те, що розлади функції печінки пов'язані з пасиреотидом.
Явища, пов'язані із серцево-судинною системою. Повідомлялося про випадки брадикардії під час застосування пасиреотиду. Ретельний моніторинг рекомендується у пацієнтів із серцевими захворюваннями та/або факторами ризику брадикардії, такими як клінічно значуща брадикардія або гострий інфаркт міокарда в анамнезі, серцеві блокади високого ступеня, застійна серцева недостатність (клас III або IV за класифікацією Нью-Йоркської кардіологічної асоціації - NYHA), нестабільна стенокардія, стійка шлуночкова тахікардія, фібриляція шлуночків. Може виникнути потреба у корекції дози лікарських засобів, наприклад, бета-блокаторів, блокаторів кальцієвих каналів або лікарських засобів, призначених для контролю електролітного балансу.
У двох дослідженнях за участю здорових добровольців було показано, що пасиреотид подовжував інтервал QT на ЕКГ. Клінічне значення такого подовження невідоме. У клінічних дослідженнях фази III у пацієнтів з акромегалією не було виявлено жодних клінічно значущих відмінностей у випадках подовження інтервалу QT між внутрішньом'язовим застосуванням пасиреотиду та аналогів соматостатину, які застосовувалися в якості активного препарату порівняння. Всі явища, пов'язані з подовження інтервалу QT, були тимчасовими та минали без терапевтичного втручання.
Епізодів тахікардії типу «пірует» не спостерігалось у жодному клінічному дослідженні пасиреотиду.
Пасиреотид слід з обережністю та з урахуванням користі/ризику застосовувати пацієнтам, які мають значний ризик розвитку подовження інтервалу QT, а саме:
- із вродженим синдромом тривалого інтервалу QT.
- з неконтрольованими або значущими серцевими захворюваннями, зокрема з недавно перенесеним інфарктом міокарда, застійною серцевою недостатністю, нестабільною стенокардією або клінічно значущою брадикардією.
- тим, хто приймає антиаритмічні лікарські засоби або інші препарати з відомою здатністю спричиняти подовження інтервалу QT.
- з гіпокаліємією та/або гіпомагніємією.
До початку терапії препаратом Сигніфор ЛАР рекомендується провести початкову ЕКГ. Бажано здійснити моніторинг щодо впливу на інтервал QTс через 21 день після початку лікування та згодом за наявності клінічних показань. Гіпокаліємію та/або гіпомагніємію необхідно відкоригувати до прийому препарату Сигніфор ЛАР і надалі проводити відповідний періодичний моніторинг під час лікування.
Гіпокортизолізм. Лікування препаратом Сигніфор ЛАР призводить до швидкого пригнічення секреції АКТГ (адренокортикотропного гормону). Під час клінічних досліджень пасиреотиду у пацієнтів з акромегалією повідомлялось про нечасті випадки гіпокортизолізму.
Таким чином, необхідно проводити моніторинг та інструктувати пацієнтів стосовно проявів та симптомів, асоційованих з гіпокортизолізмом (наприклад, слабкість, втомлюваність, анорексія, нудота, блювання, артеріальна гіпотензія, гіперкаліємія, гіпонатріємія, гіпоглікемія). У випадках підтвердженого гіпокортизолізму може виникнути потреба у тимчасовій замісній терапії із застосуванням екзогенних стероїдів (глюкокортикоїдів) та/або зменшенні дози або перерві у терапії препаратом Сигніфор ЛАР.
Жовчний міхур та пов'язані явища. Холелітіаз є встановленою небажаною реакцією, що асоціюється з тривалим застосуванням аналогів соматостатину, та про яку часто повідомлялось у клінічних дослідженнях пасиреотиду. Тому рекомендується ультразвукове обстеження жовчного міхура до та з 6- і 12-місячним інтервалами під час терапії препаратом Сигніфор ЛАР. Наявність жовчних каменів у пацієнтів, які приймають Сигніфор, перебігає переважно асимптоматично; наявність жовчних каменів з клінічними проявами слід лікувати відповідно до стандартів клінічної практики.
Гормони гіпофіза. Оскільки фармакологічна дія пасиреотиду імітує дію соматостатину, не можна виключати ймовірність інгібіції гіпофізарних гормонів, інших, ніж АКТГ. Таким чином, необхідно здійснювати моніторинг функції гіпофіза (наприклад, рівня ТТГ/вільного Т4, гормону росту/інсуліноподібного фактора росту 1) до та періодично під час терапії препаратом Сигніфор ЛАР відповідно до клінічних стандартів.
Вплив на репродуктивну функцію у жінок. Терапевтичний ефект зменшення рівнів гормону росту і нормалізації концентрації інсуліноподібного фактору росту 1 (ІФР-1) у жінок з акромегалією може потенційно відновлювати репродуктивну функцію. Пацієнткам дітородного віку слід рекомендувати використовувати відповідні засоби контрацепції, якщо необхідно, під час лікування препаратом Сигніфор ЛАР.
Розлади системи згортання крові. Пацієнти зі значно збільшеними значеннями протромбінового часу (PT) і часткового тромбопластинового часу (РТТ) або пацієнти, які отримують похідні кумарину або антикоагулянти на основі похідних гепарину, виключались з клінічних досліджень пасиреотиду, оскільки безпека поєднання з такими антикоагулянтами не була встановлена. Якщо одночасне застосування похідних кумарину або антикоагулянтів на основі похідних гепарину при внутрішньом'язовому застосуванні препарату Сигніфор ЛАР не можна уникнути, пацієнтам має бути забезпечений регулярний контроль змін їхніх параметрів коагуляції (РТ і РТТ), і дози антикоагулянту мають бути скориговані відповідним чином.
Порушення функції нирок. Через підвищення експозиції незв'язаного лікарського засобу Сигніфор ЛАР слід застосовувати з обережністю пацієнтам з тяжким порушенням функції нирок або хворобою нирок у термінальній стадії.
Вміст натрію. Цей лікарський засіб містить менше 1 ммоль натрію (23 мг) на одну рекомендовану дозу, тобто фактично він «не містить натрію».
Застосування у період вагітності або годування груддю
Вагітність
Дані щодо використання пасиреотиду у вагітних жінок обмежені. Дослідження підшкірного застосування пасиреотиду у тварин показали репродуктивну токсичність. Пасиреотид не рекомендується для застосування під час вагітності та у жінок дітородного потенціалу, які не використовують засоби контрацепції.
Годування груддю
Даних щодо екскрецію пасиреотиду у грудне молоко у людей немає. Наявні дані дослідження підшкірного застосування пасиреотиду у щурів показали, що пасиреотид проникає у молоко. Сигніфор ЛАР не слід використовувати під час годування груддю.
Фертильність
Дослідження підшкірного застосування пасиреотиду у щурів показали вплив на репродуктивні параметри у самиць. Клінічна значущість цих ефектів у людей невідома.
Здатність впливати на швидкість реакції при керуванні автотранспортом або іншими механізмами
Пасиреотид не впливає або незначною мірою впливає на здатність керувати транспортними засобами та працювати з механізмами. Пацієнтам слід порадити бути обережними при керуванні транспортними засобами або роботі з механізмами, якщо вони відчувають втому або головний біль під час лікування препаратом Сигніфор ЛАР.
Спосіб застосування та дози
Дозування.
Рекомендована початкова доза - 40 мг пасиреотиду кожні 4 тижні.
Доза може бути збільшена максимум до 60 мг для пацієнтів, у яких рівні гормону росту (ГР) та/або інсуліноподібного фактору росту 1 (ІФР-1) недостатньо контролюються через 3 місяці лікування препаратом в дозі 40 мг.
Для контролю підозрюваних побічних реакцій або надмірної реакції на лікування (ІФР-1 < нижньої межі норми) може знадобитися тимчасове зниження дози. Доза може зменшуватись тимчасово або постійно, поетапно по 20 мг.
Якщо прийом дози препарату Сигніфор ЛАР пропущено, пропущену ін'єкцію слід зробити якомога скоріше. Наступну дозу слід прийняти через 4 тижні після введення ін'єкції для відновлення графіку дозування один раз на 4 тижні.
Особливі категорії пацієнтів.
Пацієнти літнього віку ( ≥ 65 років). Дані щодо застосування препарату Сигніфор ЛАР пацієнтами віком від 65 років обмежені, але доказів, які свідчили б про необхідність корекції дози у таких пацієнтів, немає.
Порушення функції нирок. Корекція дози у пацієнтів з порушенням функції нирок не потрібна.
Порушення функції печінки. Корекція дози у пацієнтів з легким порушенням функції печінки не потрібна (клас А за Чайлд-П'ю). Рекомендована початкова доза для пацієнтів з помірним порушенням функції печінки (клас В за Чайлд-П'ю) становить 20 мг кожні 4 тижні. Максимальна рекомендована доза для таких пацієнтів становить 40 мг кожні 4 тижні. Препарат Сигніфор ЛАР не слід застосовувати пацієнтам з тяжким порушенням функції печінки (клас С за Чайлд-П'ю).
Спосіб застосування.
Препарат Сигніфор ЛАР має вводитись у формі глибокої внутрішньом'язової ін'єкції спеціально підготовленим медичним працівником. Суспензію препарату слід готувати тільки безпосередньо перед введенням.
Місце повторних внутрішньом'язових ін'єкцій слід чергувати між лівим і правим сідничним м'язом.
Компоненти набора для ін'єкцій:
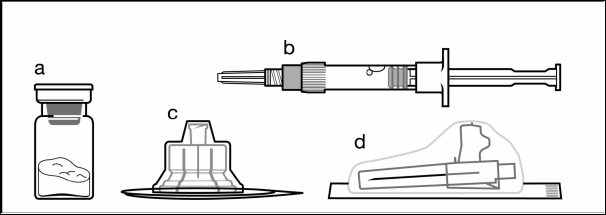
a один флакон, що містить порошок;
b один попередньо заповнений шприц з розчинником;
c один адаптер для флакона для приготування суспензії;
d одна безпечна голка для ін'єкції (20G x 1,5ʺ)
Для того щоб належним чином приготувати суспензію препарату Сигніфор ЛАР слід ретельно дотримуватися вказівок приведеним нижче.
|
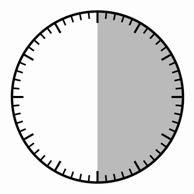
Крок 1 Дістаньте препарат Сигніфор ЛАР з холодильника. Увага Набір для ін'єкції має досягнути кімнатної температури. Слід залишити набір для ін'єкції при кімнатній температурі щонайменше на 30 хвилин (але не більше ніж на 24 години). Примітка: не використаний протягом 24 годин набір для ін'єкції можна повертати знову в холодильник. |
|
|
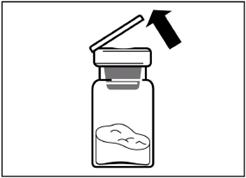
Крок 2 Зніміть з флакона пластикову кришку та протріть резинову пробку флакона спиртовою серветкою. |
|
|
Видаліть плівку з упаковки, в якій знаходиться адаптер для флакона, але НЕ виймайте його з цієї упаковки. Тримаючи адаптер для флакона за упаковку, помістіть його на верхівку флакона та проштовхніть адаптер до кінця вниз, до фіксації (характерний звук клацання). |
|
|
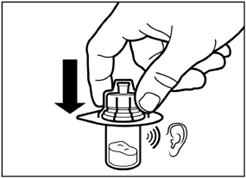
Вертикальним рухом вверх зніміть пластиковий контейнер з адаптера для флакона. |
|
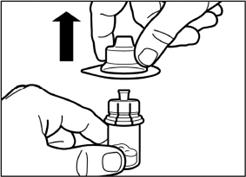
|
Крок 3 Зніміть ковпачок з попередньо заповненого шприца з розчинником, і вкрутіть шприц в адаптер для флакона |
|
|
Повільно натисніть на поршень вниз до кінцевого положення, щоб перемістити весь розчинник у флакон. |
|
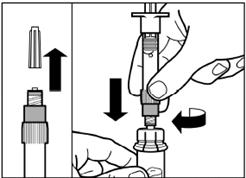
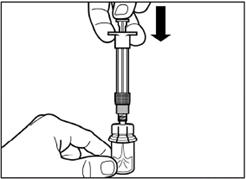
|
Крок 4 Увага: тримаючи поршень натиснутим, акуратно переміщайте флакон в горизонтальній площині протягом, щонайменше, 30 секунд доки не сформується однорідна суспензія. Якщо порошок не повністю суспендувався, знову акуратно перемішайте вміст шляхом переміщення флакона в горизонтальній площині протягом 30 секунд. |
|
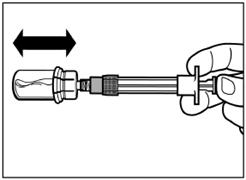
|
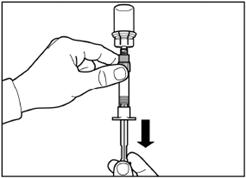
Крок 5 Переверніть флакон з приєднаним до нього шприцем догори дном, повільно потяніть поршень вниз для переміщення вмісту з флакона до шприца. |
|
|
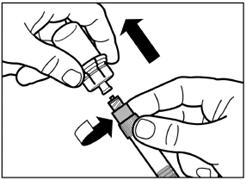
Відкрутіть шприц від адаптера для флакона. |
|
|
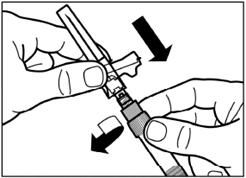
Крок 6 Накрутіть на шприц безпечну голку для ін'єкції. |
|
|
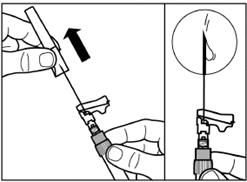
Зніміть з голки захисний ковпачок, потягнувши його вверх по лінії голки. З метою уникнення седиментації, ви можете підтримувати однорідність суспензії шляхом легкого покачування шприца. Легко постукайте по шприцу, щоб видимі пухирці повітря піднялися нагору, після чого видаліть їх обережно натискаючи на поршень. Тепер суспензія препарату готова для негайного застосування. |
|
|
Крок 7 Сигніфор ЛАР можна вводити тільки глибоко внутрішньом'язово. Підготуйте місце для ін'єкції протерши його спиртовим тампоном. Введіть голку до кінця в правий або лівий сідничний м'яз під кутом 90° до поверхні шкіри. Трохи потягніть поршень назад, щоб переконатися, що голка не потрапила в кровоносну судину (якщо голка потрапила в кровоносну судину, введіть її в інше місце). Повільно натискайте на поршень доки шприц не стане пустим. Витягніть голку із місця ін'єкції та активуйте захисний механізм (як показано на малюнку нижче). |
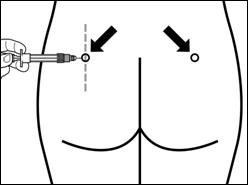
|
|
Крок 8 Активуйте захисний механізм голки одним із зазначених нижче способів: - притиснути відкидну частину захисного пристрою до твердої поверхні (малюнок A); - або натиснути вказівним пальцем на відкидну частину захисного пристрою (малюнок B). Звук клацання підтверджує правильну активацію захисного механізму. Негайно утилізуйте флакон та шприц з голкою в контейнер для гострих речей. |
|
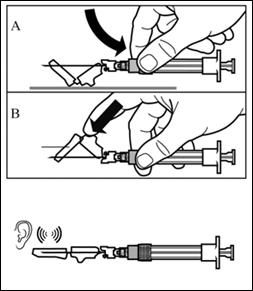
Діти. Безпека та ефективність препарату Сигніфор ЛАР у дітей та підлітків віком від 0 до 18 років не вивчалась. Дані відсутні.
Передозування.
У випадку передозування рекомендується розпочати відповідне підтримувальне лікування, що визначається клінічним станом пацієнта та проводиться до зникнення симптомів.
Побічні реакції
Резюме профілю безпеки.
Оцінку безпеки проводили у 491 пацієнта з акромегалією, які отримували пасиреотид (419 пацієнтів отримували пасиреотид внутрішньом'язово та 72 отримували пасиреотид підшкірно) під час досліджень фази I, II та III. Профіль безпеки пасиреотиду внутрішньом'язово відповідає даним щодо препаратів класу аналогів соматостатину, за винятком більш високого ступеня і частоти гіперглікемії, що спостерігались під час внутрішньом'язового застосування пасиреотиду.
Найчастішими побічними реакціями (частота ≥ 1/10) на основі зведених даних про безпеку в ході досліджень фази ІІІ C2305 і C2402 були (в порядку убування): діарея (найчастіше спостерігалась в дослідженні C2305), жовчнокам'яна хвороба, гіперглікемія (найчастіше спостерігалась в дослідженні C2402) і цукровий діабет. Побічні реакції відповідно до загальних критеріїв токсичності ступеня 3 і 4 були, в основному, пов'язані з гіперглікемією.
Зведений аналіз побічних реакцій, про які повідомлялося до дати блокування бази даних щодо досліджень C2305 та C2402, представлений нижче. Побічні реакції наведено відповідно до основних класів систем органів за Медичним словником для нормативно-правової діяльності (MedDRA). У межах кожного класу систем органів небажані реакції розподілено за частотою. У межах кожної групи за частотою небажані реакції представлені у порядку зменшення серйозності. Частоту визначено наступним чином: дуже часто (≥ 1/10); часто (від ≥ 1/100 до < 1/10); нечасто (від ≥ 1/1000 до < 1/100).
Небажані реакції (згідно з переважними термінами) при внутрішньом'язовому застосуванні пасиреотиду під час двох досліджень фази III у пацієнтів з акромегалією.
З боку крові та лімфатичної системи:
часто - анемія.
З боку ендокринної системи:
часто - недостатність надниркових залоз*.
Розлади обміну речовин та харчування:
дуже часто - гіперглікемія, цукровий діабет;
часто - цукровий діабет ІІ типу, порушення переносимості глюкози.
З боку нервової системи:
часто - головний біль, запаморочення.
З боку серця:
часто - синусова брадикардія**, пролонгація інтервалу QT.
З боку травного тракту:
дуже часто - діарея;
часто - нудота, здуття живота, болі в животі.
З боку печінки та жовчовивідних шляхів:
дуже часто - холелітіаз.
З боку шкіри та підшкірних тканин:
часто - алопеція.
Загальні розлади та реакції в місці введення препарату:
часто - реакції в місці ін'єкції***.
Дослідження:
часто - підвищення рівня глікозильованого гемоглобіну, підвищення рівня аланінамінотрансферази, підвищення рівня глюкози в крові, підвищення рівня креатинінфосфокінази в крові;
нечасто - підвищення рівня амілази.
* Недостатність надниркових залоз включає наступні визначення: адреналова недостатність і зниження рівня кортизолу в крові.
** Синусова брадикардія включає наступні визначення: брадикардія та синусова брадикардія.
*** Реакції в місці ін'єкції включають наступні визначення: біль у місці ін'єкції, вузлик у місці ін'єкції, дискомфорт у місці ін'єкції, синець у місці ін'єкції, свербіж у місці ін'єкції, реакції у місці ін'єкції та набрякання у місці ін'єкції.
Опис окремих побічних реакцій.
Розлади обміну глюкози. Підвищення рівнів глюкози натщесерце було найчастішим відхиленням лабораторних показників ступеню 3/4 під час двох досліджень фази ІІІ. У дослідженні C2305 підвищення рівнів глюкози натщесерце ступеню 3 повідомлялись у 9,7 % та 0,6 %, а ступеню 4 - у 0,6 % та 0 % пацієнтів з акромегалією, які отримували пасиреотид внутрішньом'язово та октреотид внутрішньом'язово, відповідно. У дослідженні C2402, підвищення рівнів глюкози натщесерце ступеню 3 повідомлялись у 14,3 % та 17,7 % пацієнтів з акромегалією, що отримували пасиреотид 40 мг та 60 мг внутрішньом'язово, відповідно, та у 0 % пацієнтів у групі активного контролю. Два невідкладних випадки, пов'язаних з гіперглікемією (діабетичний кетоацидоз, та діабетична гіперглікемічна кома) були зареєстровані після збільшення дози пасиреотиду до 60 мг у пацієнтів, які раніше не отримували лікування; один у пацієнта з нелікованою гіперглікемією та HbA1c > 8 % до початку застосування пасиреотиду та один у пацієнта з нелікованою гіперглікемією та плазмовим рівнем глюкози натще 359 мг/дл, відповідно. В обох дослідженнях рівні FPG та HbA1c підвищились протягом перших трьох місяців внутрішньом'язового застосування пасиреотиду. У пацієнтів, які раніше не отримували лікування (дослідження C2305) середнє абсолютне підвищення FPG та HbA1c було, здебільшого, подібним у всіх пацієнтів, які отримували пасиреотид внутрішньом'язово, незалежно від вихідних значень.
Підвищення рівнів глюкози та HbA1c у плазмі натщесерце, що спостерігались після внутрішньом'язового застосування пасиреотиду, були оборотними після відміни препарату.
Рекомендується моніторинг рівня глюкози в крові у пацієнтів, які застосовують препарат Сигніфор ЛАР.
Розлади з боку травного тракту. Під час лікування препаратом Сигніфор ЛАР часто повідомлялося про розлади з боку травного тракту. Ці явища були зазвичай низького ступеню тяжкості, не вимагали втручання та минали при продовженні лікування. Шлунково-кишкові розлади були менш частими у пацієнтів, яким не забезпечувався достатній контроль, порівняно з пацієнтами, що раніше не отримували лікування.
Реакції в місці ін'єкції. Під час досліджень фази III реакції у місці ін'єкції (наприклад, біль у місці ін'єкції, дискомфорт у місці ін'єкції) мали ступінь тяжкості 1 або 2 і були подібними при внутрішньом'язовому застосуванні пасиреотиду та внутрішньом'язовому застосуванні октреотиду. Частота таких явищ була найвищою під час перших 3 місяців лікування. Небажані явища, пов'язані з реакціями у місці ін'єкції, рідше спостерігались у пацієнтів, яким не забезпечується достатній контроль, порівняно з пацієнтами, що раніше не отримували лікування.
Подовження інтервалу QT. У дослідженні C2305 частка пацієнтів з нововиявленими помітними інтервалами QT/QTc була співставною між групами, які отримували пасиреотид внутрішньом'язово та октреотид внутрішньом'язово до перехресної фази, із кількома помітними аномальними значеннями. У жодного пацієнта значення QTcF не було > 500 мс. Значення QTcF > 480 мс спостерігалися у 3 пацієнтів порівняно з 2 пацієнтами у групі внутрішньом'язового застосування пасиреотиду та внутрішньом'язового застосування октреотиду, відповідно, та подовження QTcF > 60 мс порівняно з вихідним значенням повідомлялось у 2 пацієнтів порівняно з 1 пацієнтом у відповідних групах. У дослідженні C2402 єдиним помітним відхиленням від норми було значення QTcF > 480 мс у 1 пацієнта у групі внутрішньом'язового застосування пасиреотиду 40 мг.
Печінкові ферменти. Повідомлялося про транзиторне підвищення рівня печінкових ферментів на тлі застосування аналогів соматостатину, що також спостерігалось у пацієнтів, які приймали пасиреотид в ході клінічних досліджень. Підвищення в основному було асимптоматичним, з низьким ступенем і оборотним при продовженні лікування. Кілька випадків одночасного підвищення АЛТ більше 3 x ВМН та білірубіну більше 2 x ВМН спостерігались при підшкірному застосуванні, однак не у пацієнтів з акромегалією, які отримували пасиреотид внутрішньом'язово. Всі випадки одночасних підвищень спостерігались протягом 10 днів з моменту початку лікування. Пацієнти одужали без клінічних наслідків, а результати тестів функції печінки повернулися до вихідних значень після відміни лікування.
Рекомендується моніторинг рівня печінкових ферментів до та під час лікування препаратом Сигніфор ЛАР відповідно до клінічних стандартів.
Ферменти підшлункової залози. У пацієнтів, які приймали пасиреотид у ході клінічних досліджень, спостерігалось асимптоматичне підвищення рівня ліпази та амілази. Підвищення було переважно низького ступеня і мало оборотний характер при продовженні лікування. Панкреатит є потенційною несприятливою реакцією, пов'язаною із застосуванням аналогів соматостатину в результаті зв'язку між жовчнокам'яною хворобою та гострим панкреатитом.
Несумісність
Через відсутність досліджень сумісності цей лікарський засіб не слід змішувати з іншими лікарськими засобами.
Термін придатності. 3 роки.
Умови зберігання.
Зберігати в оригінальній упаковці при температурі 2-8 ºC. Не заморожувати. Зберігати у недоступному для дітей місці.
Упаковка. Порошок у флаконі з коричневого скла місткістю 6 мл, закупорений резиновою пробкою сірого кольору під алюмінієвим ковпачком системи flip-off сірого (для дозування 20 мг), червоного (для дозування 40 мг) або помаранчевого (для дозування 60 мг) кольору у комплекті з: розчинником у попередньо заповненому шприці місткістю 3 мл із безбарвного скла з 2 резиновими пробками сірого кольору, упором для пальців, поршнем та ковпачком; одною голкою та одним адаптером в картонній коробці.
Категорія відпуску. За рецептом.
Виробник. Новартіс Фарма Штейн АГ / Novartis Pharma Stein AG.
Місцезнаходження виробника та адреса місця провадження його діяльності.
Шаффхаусерштрассе, 4332 Штейн, Швейцарія / Schaffhauserstrasse, 4332 Stein, Switzerland.
Інші медикаменти цього ж виробника
Форма: таблетки, вкриті цукровою оболонкою, по 25 мг № 30 (10х3) у блістерах
Форма: краплі очні, 0,25 % по 5 мл у флаконах-крапельницях "Дроп-Тейнер®"; по 1 флакону-крапельниці в коробці з картону
Форма: таблетки, вкриті плівковою оболонкою, по 10 мг/160 мг/12,5 мг по 14 таблеток у блістері; по 1 або 2 блістери в картонній коробці
Форма: краплі очні, по 2,5 мл у флаконі-крапельниці; по 1 флакону-крапельниці в проміжній упаковці, що вкладається в коробку з картону
Форма: таблетки, вкриті плівковою оболонкою, 5 мг/160 мг по 14 таблеток у блістері; по 1 або по 2 блістери в коробці